General Information About Osteosarcoma and Undifferentiated Pleomorphic Sarcoma (UPS) (Formerly Called Malignant Fibrous Histiocytoma [MFH]) of Bone
Disease Overview
Osteosarcoma occurs predominantly in adolescents and young adults. Review of data from the National Cancer Institute's National Childhood Cancer Registry resulted in an estimated osteosarcoma incidence rate of 5.4 cases per 1 million each year in people aged 0 to 19 years and 4 cases per 1 million each year in people younger than 40 years.[1] The U.S. Census Bureau estimated that there were 82 million people between the ages of 0 and 19 years, resulting in an incidence of roughly 440 cases per year in this age group.
Osteosarcoma accounts for approximately 5% of childhood tumors. In children and adolescents, more than 50% of these tumors arise from the long bones around the knee. Osteosarcoma is rarely observed in soft tissue or visceral organs. There appears to be no difference in presenting symptoms, tumor location, and outcome for younger patients (<12 years) compared with adolescents.[2,3]
Two trials conducted in the 1980s were designed to determine whether chemotherapy altered the natural history of osteosarcoma after surgical removal of the primary tumor. The outcome of these patients recapitulated the historical experience before 1970. More than one-half of these patients developed metastases within 6 months of diagnosis, and overall, approximately 90% developed recurrent disease within 2 years of diagnosis.[4] Overall survival (OS) for patients treated with surgery alone was statistically inferior.[5] The natural history of osteosarcoma has not changed over time, and fewer than 20% of patients with localized, resectable primary tumors treated with surgery alone can be expected to survive free of relapse.[4,6]; [7][Level of evidence A1]
In 2013, the World Health Organization (WHO) published an update to the Classification of Tumors of Soft Tissue and Bone.[8] They removed the term malignant fibrous histiocytoma (MFH) and replaced it with undifferentiated pleomorphic sarcoma (UPS). This type of sarcoma is much more common in soft tissues. However, it does arise in bone. In bone, it has features that are histologically similar to osteosarcoma, but it does not produce osteoid. Most of the literature describing the clinical behavior and response to therapy for this histology in bone was published before the 2013 WHO update, and a search for UPS of bone will not retrieve these articles. The citations in this summary appear with their titles as published. Therefore, many references will describe MFH of bone, a condition now called UPS of bone.
Diagnostic Evaluation
Osteosarcoma can be diagnosed by core needle biopsy or open surgical biopsy. It is preferable that the biopsy be performed by a surgeon skilled in the techniques of limb sparing (removal of the malignant bone tumor without amputation and replacement of bones or joints with allografts or prosthetic devices). In these cases, the original biopsy incision placement is crucial. Inappropriate alignment of the biopsy or inadvertent contamination of soft tissues can render subsequent limb-preserving reconstructive surgery impossible.
Prognostic Factors
Dramatic improvements in survival have been achieved for children and adolescents with cancer. Between 1975 and 2020, childhood cancer mortality decreased by more than 50%.[1,9,10] For osteosarcoma, the 5-year relative survival rate increased over the same time from 40% to 72% in children younger than 15 years and from 56% to approximately 71% in adolescents aged 15 to 19 years. However, there has been no substantial improvement since the 1980s.[1,11]
In general, prognostic factors for osteosarcoma have not been helpful in identifying patients who might benefit from treatment intensification or who might require less therapy while maintaining an excellent outcome.
Factors that may influence outcome include the following:[12]
- Primary tumor site.
- Size of the primary tumor.
- Presence of clinically detectable metastatic disease.
- Surgical resectability of primary tumor.
- Degree of tumor necrosis after administration of neoadjuvant chemotherapy.
- Age and sex.
- Other possible prognostic factors.
Primary tumor site
The site of the primary tumor is a significant prognostic factor for patients with localized disease. Among extremity tumors, distal sites have a more favorable prognosis than do proximal sites. Axial skeleton primary tumors are associated with the greatest risk of progression and death, primarily related to the inability to achieve a complete surgical resection.
Prognostic considerations for the axial skeleton and extraskeletal sites are as follows:
- Pelvis: Pelvic osteosarcomas make up 7% to 9% of all osteosarcomas. Survival rates for patients with pelvic primary tumors are 20% to 47%.[13,14,15] Complete surgical resection is associated with a positive outcome for osteosarcoma of the pelvis in some cohorts of patients.[13,16]
- Craniofacial/head and neck: In patients with craniofacial osteosarcoma, those with primary sites in the mandible and maxilla have a better prognosis than do patients with other primary sites in the head and neck.[17,18,19] For patients with osteosarcoma of craniofacial bones, complete resection of the primary tumor with negative margins is essential for cure.[20,21,22] When treated with surgery alone, patients who have osteosarcoma of the head and neck have a better prognosis than those who have appendicular lesions. However, surgery alone without adjuvant therapy is not recommended for high-grade osteosarcoma of the head and neck.
Despite a relatively high rate of inferior necrosis after neoadjuvant chemotherapy, fewer patients with craniofacial primary tumors develop systemic metastases than do patients with osteosarcoma originating in the extremities. This may be the result of the inclusion of patients with lower-grade tumors in the cohorts reported.[23,24,25]
A meta-analysis concluded that systemic adjuvant chemotherapy improves the prognosis for patients with osteosarcoma of the head and neck, while small series have not shown such a benefit.[23,24,25] Another large meta-analysis detected no benefit of chemotherapy for patients with osteosarcoma of the head and neck, but suggested that incorporating chemotherapy into the treatment plan for patients with high-grade tumors may improve survival.[22] A retrospective analysis identified a trend toward better survival in patients with high-grade osteosarcoma of the mandible and maxilla who received adjuvant chemotherapy.[22,26]
Radiation therapy was found to improve local control, disease-specific survival, and OS in a retrospective study of patients with osteosarcoma of the craniofacial bones who had positive or uncertain margins after surgical resection.[27][Level of evidence C1] Radiation-associated craniofacial osteosarcomas are generally high-grade lesions, usually fibroblastic, and tend to recur locally with a high rate of metastasis.[28]
- Extraskeletal: Osteosarcoma in extraskeletal sites is rare in children and young adults. With current combined-modality therapy, the outcome of patients with extraskeletal osteosarcoma appears to be similar to that of patients with primary tumors of bone.[29]
Size of the primary tumor
In some series, patients with larger tumors appeared to have a worse prognosis than patients with smaller tumors.[12,30,31] Tumor size has been assessed by longest single dimension, cross-sectional area, or estimate of tumor volume; all assessments have correlated with outcome.
Elevated serum lactate dehydrogenase (LDH), which also correlates with poorer outcome, is a likely surrogate for tumor volume.[14]
Presence of clinically detectable metastatic disease
Patients with localized disease have a much better prognosis than patients with overt metastatic disease. As many as 20% of patients have radiographically detectable metastases at diagnosis, with the lung being the most common site.[32] The prognosis for patients with metastatic disease appears to be determined largely by site(s) of metastases, number of metastases, and surgical resectability of the metastatic disease.[33,34]
- Site of metastases: Prognosis appears more favorable for patients with fewer pulmonary nodules and for those with unilateral rather than bilateral pulmonary metastases.[33] Not all patients with suspected pulmonary metastases at diagnosis have osteosarcoma confirmed at the time of lung resection. In one large series, approximately 25% of patients had exclusively benign lesions removed at the time of surgery.[34]
- Number of metastases: Patients with skip metastases (at least two discontinuous lesions in the same bone) have been reported to have inferior prognoses.[35] However, an analysis of the German Cooperative Osteosarcoma Study suggests that skip lesions in the same bone do not confer an inferior prognosis if they are included in planned surgical resection. Skip metastasis in a bone other than the primary bone should be considered systemic metastasis.[36]
Historically, metastasis across a joint was referred to as a skip lesion, but subsequent classification by the American Joint Committee on Cancer excluded such lesions as skip lesions.[37] They might be considered hematogenous spread and have a worse prognosis.[36]
Patients with multifocal osteosarcoma (defined as multiple bone lesions without a clear primary tumor) have an extremely poor prognosis.[38,39]
- Surgical resectability of metastases: Patients who have complete surgical ablation of the primary and metastatic tumor (when confined to the lung) after chemotherapy may attain long-term survival, although overall event-free survival (EFS) rates remain about 20% to 30% for patients with metastatic disease at diagnosis.[33,34,40,41] Patients with metastatic osteosarcoma were eligible for the European and American Osteosarcoma Study (EURAMOS) only if they had disease that was potentially resectable. Although the patients with metastatic disease had an overall 5-year EFS rate of only 28%, those who achieved a complete surgical remission at all sites (3–6 months after diagnosis) had a 5-year EFS rate of 64% and an OS rate of 79%.[31]
Surgical resectability of the primary tumor
Resectability of the tumor is a critical prognostic feature. Complete resection of the primary tumor and any skip lesions with adequate margins is generally considered essential for cure. For patients with axial skeletal primary tumors who either do not undergo surgery for their primary tumor or who undergo surgery that results in positive margins, radiation therapy may improve survival.[16,42]
A retrospective review of patients with craniofacial osteosarcoma performed by the cooperative German-Austrian-Swiss osteosarcoma study group reported that incomplete surgical resection was associated with inferior survival probability.[17][Level of evidence C1] In a European cooperative study, the size of the margin was not significant. However, prognosis was better when both the biopsy and resection were performed at a center with orthopedic oncology experience.[14]
Degree of tumor necrosis after neoadjuvant chemotherapy
Most treatment protocols for osteosarcoma use an initial period of systemic chemotherapy before definitive resection of the primary tumor (or resection of sites of metastases). The pathologist assesses necrosis in the resected tumor. Patients with at least 90% necrosis in the primary tumor after induction chemotherapy have a better prognosis than do patients with less necrosis.[30] Patients with less necrosis (<90%) in the primary tumor after initial chemotherapy have a higher rate of recurrence within the first 2 years than do patients with a more favorable amount of necrosis (≥90%).[43]
Less necrosis should not be interpreted to mean that chemotherapy has been ineffective. Cure rates for patients with little or no necrosis after induction chemotherapy are much higher than cure rates for patients who receive no chemotherapy. The EFS rate for patients who receive no adjuvant chemotherapy is approximately 11%.[44] Many large published series of patients treated with chemotherapy have reported EFS rates of 40% to 50% for patients with little or no necrosis in the primary tumor after initial systemic chemotherapy.[45,46,47] A review of two consecutive prospective trials performed by the Children's Oncology Group showed that histological necrosis in the primary tumor after initial chemotherapy was affected by the duration and intensity of the initial period of chemotherapy. More necrosis was associated with better outcome in both trials, but the magnitude of the difference between patients with more and less necrosis was diminished with a longer and more intensive period of initial chemotherapy.[48][Level of evidence B1]
Age and sex
Patients in the older adolescent and young adult age group, typically defined as age 18 to 40 years, tend to have a worse prognosis. In addition, male sex has been associated with a worse prognosis.[31,49,50] Compared with the other prognostic factors listed, both age and sex have a relatively minor impact on outcome.
Other possible prognostic factors
Other factors that may be prognostic but with either limited or conflicting data include the following:
- Subsequent neoplasms. Patients with osteosarcoma as a subsequent neoplasm, including tumors arising in a radiation field, share the same prognosis as patients with de novo osteosarcoma if they are treated aggressively with complete surgical resection and multiagent chemotherapy.[51,52,53,54]
In a German series, approximately 25% of patients with craniofacial osteosarcoma had osteosarcoma as a second tumor, and in 8 of these 13 patients, osteosarcoma arose after treatment for retinoblastoma. In this series, there was no difference in outcome for primary or secondary craniofacial osteosarcoma.[17]
- Laboratory abnormalities. Possible prognostic factors identified for patients with conventional localized high-grade osteosarcoma include LDH level, alkaline phosphatase level, and histological subtype.[30,46,49,55,56,57,58]
- Body mass index. Higher body mass index at initial presentation is associated with worse OS.[59]
- Pathological fracture. Some studies have suggested that a pathological fracture at diagnosis or during preoperative chemotherapy does not have adverse prognostic significance.[6]; [60,61][Level of evidence C1]; [62][Level of evidence C2]
However, a systematic review of nine cohort studies examined the impact of pathological fractures on outcome in patients with osteosarcoma. The review included 2,187 patients, 311 of whom had a pathological fracture. The presence of a pathological fracture correlated with decreased EFS and OS.[63] In two additional series, a pathological fracture at diagnosis was associated with a worse overall outcome.[64]; [65][Level of evidence C1] A retrospective analysis of 2,847 patients with osteosarcoma from the German cooperative group identified 321 patients (11.3%) with a pathological fracture before the initiation of systemic therapy.[66][Level of evidence C1] In pediatric patients, OS and EFS did not differ significantly between patients with and without a pathological fracture. In adults, the 5-year OS rate in patients with a pathological fracture was 46% versus 69% for patients without a pathological fracture (P < .001). The 5-year EFS rate in adults was 36% for patients with a pathological fracture versus 56% for patients without a pathological fracture (P < .001). In a multivariable analysis, the presence of a pathological fracture was not a statistically significant factor for OS or EFS in the total cohort or in pediatric patients. In adult patients, presence of a pathological fracture remained an independent prognostic factor for OS (hazard ratio, 1.893; P = .013).
- Time to definitive surgery. In a large series, a delay of 21 days or longer from the time of definitive surgery to the resumption of chemotherapy was an adverse prognostic factor.[67]
- Genetic factors.
- ERBB2 expression. There are conflicting data concerning the prognostic significance of this human epidermal growth factor.[68,69,70]
- Tumor cell ploidy.[71]
- Specific chromosomal gains or losses.[72]
- Loss of heterozygosity of the RB1 gene.[73,74]
- Loss of heterozygosity of the TP53 locus.[75]
- Increased expression of p-glycoprotein.[76,77] A prospective analysis of p-glycoprotein expression determined by immunohistochemistry failed to identify prognostic significance for patients with newly diagnosed osteosarcoma, although earlier studies suggested that overexpression of p-glycoprotein predicted poor outcome.[78]
For more information, see the Genomics of Osteosarcoma section.
References:
-
National Cancer Institute: NCCR*Explorer: An interactive website for NCCR cancer statistics. Bethesda, MD: National Cancer Institute. Available online. Last accessed December 15, 2023.
-
Bacci G, Longhi A, Bertoni F, et al.: Primary high-grade osteosarcoma: comparison between preadolescent and older patients. J Pediatr Hematol Oncol 27 (3): 129-34, 2005.
-
Bacci G, Balladelli A, Palmerini E, et al.: Neoadjuvant chemotherapy for osteosarcoma of the extremities in preadolescent patients: the Rizzoli Institute experience. J Pediatr Hematol Oncol 30 (12): 908-12, 2008.
-
Link MP, Goorin AM, Miser AW, et al.: The effect of adjuvant chemotherapy on relapse-free survival in patients with osteosarcoma of the extremity. N Engl J Med 314 (25): 1600-6, 1986.
-
Link MP: The multi-institutional osteosarcoma study: an update. Cancer Treat Res 62: 261-7, 1993.
-
Bacci G, Ferrari S, Longhi A, et al.: Nonmetastatic osteosarcoma of the extremity with pathologic fracture at presentation: local and systemic control by amputation or limb salvage after preoperative chemotherapy. Acta Orthop Scand 74 (4): 449-54, 2003.
-
Bernthal NM, Federman N, Eilber FR, et al.: Long-term results (>25 years) of a randomized, prospective clinical trial evaluating chemotherapy in patients with high-grade, operable osteosarcoma. Cancer 118 (23): 5888-93, 2012.
-
Fletcher CDM, Bridge JA, Hogendoorn P, et al., eds.: WHO Classification of Tumours of Soft Tissue and Bone. 4th ed. IARC Press, 2013.
-
Smith MA, Altekruse SF, Adamson PC, et al.: Declining childhood and adolescent cancer mortality. Cancer 120 (16): 2497-506, 2014.
-
Surveillance Research Program, National Cancer Institute: SEER*Explorer: An interactive website for SEER cancer statistics. Bethesda, MD: National Cancer Institute. Available online. Last accessed March 6, 2024.
-
Isakoff MS, Bielack SS, Meltzer P, et al.: Osteosarcoma: Current Treatment and a Collaborative Pathway to Success. J Clin Oncol 33 (27): 3029-35, 2015.
-
Pakos EE, Nearchou AD, Grimer RJ, et al.: Prognostic factors and outcomes for osteosarcoma: an international collaboration. Eur J Cancer 45 (13): 2367-75, 2009.
-
Donati D, Giacomini S, Gozzi E, et al.: Osteosarcoma of the pelvis. Eur J Surg Oncol 30 (3): 332-40, 2004.
-
Andreou D, Bielack SS, Carrle D, et al.: The influence of tumor- and treatment-related factors on the development of local recurrence in osteosarcoma after adequate surgery. An analysis of 1355 patients treated on neoadjuvant Cooperative Osteosarcoma Study Group protocols. Ann Oncol 22 (5): 1228-35, 2011.
-
Isakoff MS, Barkauskas DA, Ebb D, et al.: Poor survival for osteosarcoma of the pelvis: a report from the Children's Oncology Group. Clin Orthop Relat Res 470 (7): 2007-13, 2012.
-
Ozaki T, Flege S, Kevric M, et al.: Osteosarcoma of the pelvis: experience of the Cooperative Osteosarcoma Study Group. J Clin Oncol 21 (2): 334-41, 2003.
-
Jasnau S, Meyer U, Potratz J, et al.: Craniofacial osteosarcoma Experience of the cooperative German-Austrian-Swiss osteosarcoma study group. Oral Oncol 44 (3): 286-94, 2008.
-
Laskar S, Basu A, Muckaden MA, et al.: Osteosarcoma of the head and neck region: lessons learned from a single-institution experience of 50 patients. Head Neck 30 (8): 1020-6, 2008.
-
Kassir RR, Rassekh CH, Kinsella JB, et al.: Osteosarcoma of the head and neck: meta-analysis of nonrandomized studies. Laryngoscope 107 (1): 56-61, 1997.
-
Patel SG, Meyers P, Huvos AG, et al.: Improved outcomes in patients with osteogenic sarcoma of the head and neck. Cancer 95 (7): 1495-503, 2002.
-
Smith RB, Apostolakis LW, Karnell LH, et al.: National Cancer Data Base report on osteosarcoma of the head and neck. Cancer 98 (8): 1670-80, 2003.
-
Fernandes R, Nikitakis NG, Pazoki A, et al.: Osteogenic sarcoma of the jaw: a 10-year experience. J Oral Maxillofac Surg 65 (7): 1286-91, 2007.
-
Smeele LE, Kostense PJ, van der Waal I, et al.: Effect of chemotherapy on survival of craniofacial osteosarcoma: a systematic review of 201 patients. J Clin Oncol 15 (1): 363-7, 1997.
-
Ha PK, Eisele DW, Frassica FJ, et al.: Osteosarcoma of the head and neck: a review of the Johns Hopkins experience. Laryngoscope 109 (6): 964-9, 1999.
-
Duffaud F, Digue L, Baciuchka-Palmaro M, et al.: Osteosarcomas of flat bones in adolescents and adults. Cancer 88 (2): 324-32, 2000.
-
Canadian Society of Otolaryngology-Head and Neck Surgery Oncology Study Group: Osteogenic sarcoma of the mandible and maxilla: a Canadian review (1980-2000). J Otolaryngol 33 (3): 139-44, 2004.
-
Guadagnolo BA, Zagars GK, Raymond AK, et al.: Osteosarcoma of the jaw/craniofacial region: outcomes after multimodality treatment. Cancer 115 (14): 3262-70, 2009.
-
McHugh JB, Thomas DG, Herman JM, et al.: Primary versus radiation-associated craniofacial osteosarcoma: Biologic and clinicopathologic comparisons. Cancer 107 (3): 554-62, 2006.
-
Goldstein-Jackson SY, Gosheger G, Delling G, et al.: Extraskeletal osteosarcoma has a favourable prognosis when treated like conventional osteosarcoma. J Cancer Res Clin Oncol 131 (8): 520-6, 2005.
-
Bielack SS, Kempf-Bielack B, Delling G, et al.: Prognostic factors in high-grade osteosarcoma of the extremities or trunk: an analysis of 1,702 patients treated on neoadjuvant cooperative osteosarcoma study group protocols. J Clin Oncol 20 (3): 776-90, 2002.
-
Smeland S, Bielack SS, Whelan J, et al.: Survival and prognosis with osteosarcoma: outcomes in more than 2000 patients in the EURAMOS-1 (European and American Osteosarcoma Study) cohort. Eur J Cancer 109: 36-50, 2019.
-
Meyers PA, Schwartz CL, Krailo M, et al.: Osteosarcoma: a randomized, prospective trial of the addition of ifosfamide and/or muramyl tripeptide to cisplatin, doxorubicin, and high-dose methotrexate. J Clin Oncol 23 (9): 2004-11, 2005.
-
Harris MB, Gieser P, Goorin AM, et al.: Treatment of metastatic osteosarcoma at diagnosis: a Pediatric Oncology Group Study. J Clin Oncol 16 (11): 3641-8, 1998.
-
Bacci G, Rocca M, Salone M, et al.: High grade osteosarcoma of the extremities with lung metastases at presentation: treatment with neoadjuvant chemotherapy and simultaneous resection of primary and metastatic lesions. J Surg Oncol 98 (6): 415-20, 2008.
-
Sajadi KR, Heck RK, Neel MD, et al.: The incidence and prognosis of osteosarcoma skip metastases. Clin Orthop Relat Res (426): 92-6, 2004.
-
Kager L, Zoubek A, Kastner U, et al.: Skip metastases in osteosarcoma: experience of the Cooperative Osteosarcoma Study Group. J Clin Oncol 24 (10): 1535-41, 2006.
-
American Joint Committee on Cancer: AJCC Cancer Staging Manual. 6th ed. Springer, 2002.
-
Bacci G, Fabbri N, Balladelli A, et al.: Treatment and prognosis for synchronous multifocal osteosarcoma in 42 patients. J Bone Joint Surg Br 88 (8): 1071-5, 2006.
-
Longhi A, Fabbri N, Donati D, et al.: Neoadjuvant chemotherapy for patients with synchronous multifocal osteosarcoma: results in eleven cases. J Chemother 13 (3): 324-30, 2001.
-
Goorin AM, Shuster JJ, Baker A, et al.: Changing pattern of pulmonary metastases with adjuvant chemotherapy in patients with osteosarcoma: results from the multiinstitutional osteosarcoma study. J Clin Oncol 9 (4): 600-5, 1991.
-
Bacci G, Mercuri M, Longhi A, et al.: Grade of chemotherapy-induced necrosis as a predictor of local and systemic control in 881 patients with non-metastatic osteosarcoma of the extremities treated with neoadjuvant chemotherapy in a single institution. Eur J Cancer 41 (14): 2079-85, 2005.
-
DeLaney TF, Park L, Goldberg SI, et al.: Radiotherapy for local control of osteosarcoma. Int J Radiat Oncol Biol Phys 61 (2): 492-8, 2005.
-
Kim MS, Cho WH, Song WS, et al.: time dependency of prognostic factors in patients with stage II osteosarcomas. Clin Orthop Relat Res 463: 157-65, 2007.
-
Link MP, Goorin AM, Horowitz M, et al.: Adjuvant chemotherapy of high-grade osteosarcoma of the extremity. Updated results of the Multi-Institutional Osteosarcoma Study. Clin Orthop (270): 8-14, 1991.
-
Winkler K, Beron G, Delling G, et al.: Neoadjuvant chemotherapy of osteosarcoma: results of a randomized cooperative trial (COSS-82) with salvage chemotherapy based on histological tumor response. J Clin Oncol 6 (2): 329-37, 1988.
-
Meyers PA, Heller G, Healey J, et al.: Chemotherapy for nonmetastatic osteogenic sarcoma: the Memorial Sloan-Kettering experience. J Clin Oncol 10 (1): 5-15, 1992.
-
Bielack S, Jürgens H, Jundt G, et al.: Osteosarcoma: the COSS experience. Cancer Treat Res 152: 289-308, 2009.
-
Bishop MW, Chang YC, Krailo MD, et al.: Assessing the Prognostic Significance of Histologic Response in Osteosarcoma: A Comparison of Outcomes on CCG-782 and INT0133-A Report From the Children's Oncology Group Bone Tumor Committee. Pediatr Blood Cancer 63 (10): 1737-43, 2016.
-
Janeway KA, Barkauskas DA, Krailo MD, et al.: Outcome for adolescent and young adult patients with osteosarcoma: a report from the Children's Oncology Group. Cancer 118 (18): 4597-605, 2012.
-
Collins M, Wilhelm M, Conyers R, et al.: Benefits and adverse events in younger versus older patients receiving neoadjuvant chemotherapy for osteosarcoma: findings from a meta-analysis. J Clin Oncol 31 (18): 2303-12, 2013.
-
Bielack SS, Kempf-Bielack B, Heise U, et al.: Combined modality treatment for osteosarcoma occurring as a second malignant disease. Cooperative German-Austrian-Swiss Osteosarcoma Study Group. J Clin Oncol 17 (4): 1164, 1999.
-
Tabone MD, Terrier P, Pacquement H, et al.: Outcome of radiation-related osteosarcoma after treatment of childhood and adolescent cancer: a study of 23 cases. J Clin Oncol 17 (9): 2789-95, 1999.
-
Shaheen M, Deheshi BM, Riad S, et al.: Prognosis of radiation-induced bone sarcoma is similar to primary osteosarcoma. Clin Orthop Relat Res 450: 76-81, 2006.
-
Bacci G, Longhi A, Forni C, et al.: Neoadjuvant chemotherapy for radioinduced osteosarcoma of the extremity: The Rizzoli experience in 20 cases. Int J Radiat Oncol Biol Phys 67 (2): 505-11, 2007.
-
Bacci G, Longhi A, Versari M, et al.: Prognostic factors for osteosarcoma of the extremity treated with neoadjuvant chemotherapy: 15-year experience in 789 patients treated at a single institution. Cancer 106 (5): 1154-61, 2006.
-
Bieling P, Rehan N, Winkler P, et al.: Tumor size and prognosis in aggressively treated osteosarcoma. J Clin Oncol 14 (3): 848-58, 1996.
-
Ferrari S, Bertoni F, Mercuri M, et al.: Predictive factors of disease-free survival for non-metastatic osteosarcoma of the extremity: an analysis of 300 patients treated at the Rizzoli Institute. Ann Oncol 12 (8): 1145-50, 2001.
-
Kager L, Zoubek A, Dominkus M, et al.: Osteosarcoma in very young children: experience of the Cooperative Osteosarcoma Study Group. Cancer 116 (22): 5316-24, 2010.
-
Altaf S, Enders F, Jeavons E, et al.: High-BMI at diagnosis is associated with inferior survival in patients with osteosarcoma: a report from the Children's Oncology Group. Pediatr Blood Cancer 60 (12): 2042-6, 2013.
-
Kim MS, Lee SY, Lee TR, et al.: Prognostic effect of pathologic fracture in localized osteosarcoma: a cohort/case controlled study at a single institute. J Surg Oncol 100 (3): 233-9, 2009.
-
Xie L, Guo W, Li Y, et al.: Pathologic fracture does not influence local recurrence and survival in high-grade extremity osteosarcoma with adequate surgical margins. J Surg Oncol 106 (7): 820-5, 2012.
-
Chung LH, Wu PK, Chen CF, et al.: Pathological fractures in predicting clinical outcomes for patients with osteosarcoma. BMC Musculoskelet Disord 17 (1): 503, 2016.
-
Sun L, Li Y, Zhang J, et al.: Prognostic value of pathologic fracture in patients with high grade localized osteosarcoma: a systemic review and meta-analysis of cohort studies. J Orthop Res 33 (1): 131-9, 2015.
-
Bramer JA, Abudu AA, Grimer RJ, et al.: Do pathological fractures influence survival and local recurrence rate in bony sarcomas? Eur J Cancer 43 (13): 1944-51, 2007.
-
Schlegel M, Zeumer M, Prodinger PM, et al.: Impact of Pathological Fractures on the Prognosis of Primary Malignant Bone Sarcoma in Children and Adults: A Single-Center Retrospective Study of 205 Patients. Oncology 94 (6): 354-362, 2018.
-
Kelley LM, Schlegel M, Hecker-Nolting S, et al.: Pathological Fracture and Prognosis of High-Grade Osteosarcoma of the Extremities: An Analysis of 2,847 Consecutive Cooperative Osteosarcoma Study Group (COSS) Patients. J Clin Oncol 38 (8): 823-833, 2020.
-
Imran H, Enders F, Krailo M, et al.: Effect of time to resumption of chemotherapy after definitive surgery on prognosis for non-metastatic osteosarcoma. J Bone Joint Surg Am 91 (3): 604-12, 2009.
-
Gorlick R, Huvos AG, Heller G, et al.: Expression of HER2/erbB-2 correlates with survival in osteosarcoma. J Clin Oncol 17 (9): 2781-8, 1999.
-
Onda M, Matsuda S, Higaki S, et al.: ErbB-2 expression is correlated with poor prognosis for patients with osteosarcoma. Cancer 77 (1): 71-8, 1996.
-
Kilpatrick SE, Geisinger KR, King TS, et al.: Clinicopathologic analysis of HER-2/neu immunoexpression among various histologic subtypes and grades of osteosarcoma. Mod Pathol 14 (12): 1277-83, 2001.
-
Look AT, Douglass EC, Meyer WH: Clinical importance of near-diploid tumor stem lines in patients with osteosarcoma of an extremity. N Engl J Med 318 (24): 1567-72, 1988.
-
Ozaki T, Schaefer KL, Wai D, et al.: Genetic imbalances revealed by comparative genomic hybridization in osteosarcomas. Int J Cancer 102 (4): 355-65, 2002.
-
Feugeas O, Guriec N, Babin-Boilletot A, et al.: Loss of heterozygosity of the RB gene is a poor prognostic factor in patients with osteosarcoma. J Clin Oncol 14 (2): 467-72, 1996.
-
Heinsohn S, Evermann U, Zur Stadt U, et al.: Determination of the prognostic value of loss of heterozygosity at the retinoblastoma gene in osteosarcoma. Int J Oncol 30 (5): 1205-14, 2007.
-
Goto A, Kanda H, Ishikawa Y, et al.: Association of loss of heterozygosity at the p53 locus with chemoresistance in osteosarcomas. Jpn J Cancer Res 89 (5): 539-47, 1998.
-
Serra M, Pasello M, Manara MC, et al.: May P-glycoprotein status be used to stratify high-grade osteosarcoma patients? Results from the Italian/Scandinavian Sarcoma Group 1 treatment protocol. Int J Oncol 29 (6): 1459-68, 2006.
-
Pakos EE, Ioannidis JP: The association of P-glycoprotein with response to chemotherapy and clinical outcome in patients with osteosarcoma. A meta-analysis. Cancer 98 (3): 581-9, 2003.
-
Schwartz CL, Gorlick R, Teot L, et al.: Multiple drug resistance in osteogenic sarcoma: INT0133 from the Children's Oncology Group. J Clin Oncol 25 (15): 2057-62, 2007.
Genomics of Osteosarcoma
Molecular Features of Osteosarcoma
The genomic landscape of osteosarcoma is distinct from that of other childhood cancers. Compared with many adult cancers, it is characterized by an exceptionally high number of structural variants with a relatively small number of single nucleotide variants.[1,2]
Key observations regarding the genomic landscape of osteosarcoma include the following:
- The number of structural variants observed for osteosarcoma is high, at more than 200 structural variants per genome.[1,2] Thus, osteosarcoma has the most chaotic genome among childhood cancers. The Circos plots shown in Figure 1 illustrate the exceptionally high number of intra- and inter-chromosomal translocations that typify osteosarcoma genomes.
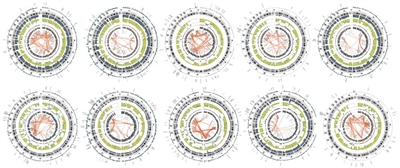
Figure 1. Circos plots of osteosarcoma cases from the National Cancer Institute's Therapeutically Applicable Research to Generate Effective Treatments (TARGET) project. The red lines in the interior circle connect chromosome regions involved in either intra- or inter-chromosomal translocations. Osteosarcoma is distinctive from other childhood cancers because it has a large number of intra- and inter-chromosomal translocations. Credit: National Cancer Institute.
- The tumor mutational burden (TMB) for children and adolescents with osteosarcoma is approximately 2 mutations per megabase and is higher than that of some other childhood cancers (e.g., Ewing sarcoma and rhabdoid tumors).[1,2] However, this rate is well below that for adult cancers such as melanoma and non-small cell lung cancer, which are responsive to checkpoint inhibitors.
- Rather than activating variants in oncogenes and inactivating variants in tumor suppressor genes, as observed in many cancer types, the genomic landscape for osteosarcoma is driven by copy number gain/amplification in chromosome regions that include oncogenes and copy number loss (deletions) in chromosome regions that include tumor suppressor genes. Recurring copy number gains and losses that affect known oncogenes and tumor suppressor genes, respectively, are described below.
Estimates of the frequency of specific genomic alterations in osteosarcoma vary from report to report. This finding could be a result of different definitions being used to define copy number alterations, different methods being used for their detection, or differences in tumor biology across patient populations (e.g., newly diagnosed versus relapsed, localized versus metastatic, or pediatric versus adult).
- Genomic alterations in TP53, leading to loss of TP53 function, are present in most osteosarcoma cases.[1] A distinctive form of TP53 inactivation occurs through structural variations in the first intron of TP53 that lead to disruption of the TP53 gene.[1] Other mechanisms of TP53 inactivation are also observed, including missense and nonsense variants and deletions of the TP53 gene.[1,2] The combination of these various mechanisms for loss of TP53 function leads to its biallelic inactivation in most cases of osteosarcoma. Because many of the structural variations leading to TP53 inactivation are best detected through whole-genome sequencing, results based on clinical genomic testing panels may show lower rates of TP53 alterations because they do not detect these changes.[3]
- MDM2 amplification, which is another genomic alteration that leads to loss of TP53 function, is observed in a minority of osteosarcoma cases (approximately 5%).[1,2,3,4]
- RB1 is commonly inactivated in osteosarcoma, sometimes by deleterious variants but more commonly by chromosomal deletion of the chromosome 13q14 region that includes RB1.[1,2,5]
- Chromosomal deletions involving chromosome 9p21 lead to CDN2A deletion in approximately 20% of osteosarcoma cases.[1,2,5]
- Among tumor oncogenes, MYC at chromosome 8q24 shows gain/amplification in approximately 10% of patients.[3,5,6] In one study, MYC gain/amplification appeared to be associated with inferior prognosis. In a second study, MYC gain/amplification was enriched in children, compared with adults.[6]
- CCNE1 at chromosome 19q12 is another tumor oncogene that shows gain/amplification in approximately 10% of patients.[3,5,6] Other oncogene-containing chromosomal regions showing chromosomal gain/amplification in a minority of osteosarcoma cases include the CDK4-harboring region at chromosome 12q14,[4,5,7] the VEGFA- and CCND3-harboring regions at chromosome 6p12,[3,4,5,7] the CCND1-harboring region at chromosome 11q13,[4] and the PDGFRA-, KIT-, and KDR-harboring regions at chromosome 4q12.[3,4,5]
- Alternative lengthening of telomeres (ALT) is the telomere maintenance mechanism employed by the majority of osteosarcoma tumors.[1,8,9]ATRX inactivating variants and gene deletions are associated with the ALT telomere maintenance mechanism. ATRX genomic alterations are present in a subset of osteosarcoma tumors that use this telomere maintenance mechanism.[1,3,9]
- Many of the genomic alterations reported for osteosarcoma tumors at diagnosis do not provide obvious therapeutic targets, as they reflect loss of tumor suppressor genes (e.g., TP53, RB1, PTEN) rather than activation of targetable oncogenes. In addition, there has been limited success across cancer diagnoses in using gains/amplifications of the oncogenes relevant to osteosarcoma to identify patients that may benefit from targeted therapy.
Genetic predisposition to osteosarcoma
Germline variants in several genes are associated with susceptibility to osteosarcoma. Table 1 summarizes the syndromes and associated genes for these conditions. A recent multi-institutional genomic study of more than 1,200 patients with osteosarcoma revealed pathogenic or likely pathogenic germline variants in autosomal dominant cancer-susceptibility genes in 18% of patients. The frequency of these cancer-susceptibility genes was higher in children aged 10 years or younger.[10]
TP53variants
Variants in TP53 are the most common germline alterations associated with osteosarcoma. Variants in this gene are found in approximately 70% of patients with Li-Fraumeni syndrome (LFS), which is associated with increased risk of osteosarcoma, breast cancer, various brain cancers, soft tissue sarcomas, and other cancers. While rhabdomyosarcoma is the most common sarcoma arising in patients aged 5 years and younger with TP53-associated LFS, osteosarcoma is the most common sarcoma in children and adolescents aged 6 to 19 years.[11] One study observed a high frequency of young patients (age <30 years) with osteosarcoma carrying a known LFS-associated or likely LFS-associated TP53 variant (3.8%) or rare exonic TP53 variant (5.7%), with an overall TP53 variant frequency of 9.5%.[12] Other groups have reported lower rates (3%–7%) of TP53 germline variants in patients with osteosarcoma.[10,13,14]
RECQL4variants
Investigators analyzed whole-exome sequencing from the germline of 4,435 pediatric cancer patients at the St. Jude Children's Research Hospital and 1,127 patients from the National Cancer Institute's Therapeutically Applicable Research to Generate Effective Treatment (TARGET) database. They identified 24 patients (0.43%) who harbored loss-of-function RECQL4 variants, including 5 of 249 patients (2.0%) with osteosarcoma.[15] These RECQL4 variants were significantly overrepresented in children with osteosarcoma, the cancer most frequently observed in patients with Rothmund-Thomson syndrome, compared with 134,187 noncancer controls in the Genome Aggregation Database (gnomAD v2.1; P = .00087; odds ratio, 7.1; 95% confidence interval, 2.9–17). Nine of the 24 individuals (38%) possessed the same c.1573delT (p.Cys525Alafs) variant located in the highly conserved DNA helicase domain, suggesting that disruption of this domain is central to oncogenesis.
Table 1. Genetic Diseases That Predispose to Osteosarcomaa
| Syndrome |
Description |
Location |
Gene |
Function |
| AML = acute myeloid leukemia; IL-1 = interleukin-1; MDS = myelodysplastic syndrome; RANKL = receptor activator of nuclear factor kappa beta ligand; TNF = tumor necrosis factor. |
| a Adapted from Kansara et al.[16] |
| Bloom syndrome[17] |
Rare inherited disorder characterized by short stature and sun-sensitive skin changes. Often presents with a long, narrow face, small lower jaw, large nose, and prominent ears. |
15q26.1 |
BLM |
DNA helicase |
| Diamond-Blackfan anemia[18] |
Inherited pure red cell aplasia. Patients at risk for MDS and AML. Associated with skeletal abnormalities such as abnormal facial features (flat nasal bridge, widely spaced eyes). |
|
Ribosomal proteins |
Ribosome production[18,19] |
| Li-Fraumeni syndrome[20] |
Inherited variant inTP53gene. Affected family members at increased risk of bone tumors, breast cancer, leukemia, brain tumors, and sarcomas. |
17p13.1 |
TP53 |
DNA damage response |
| Paget disease[21] |
Excessive breakdown of bone with abnormal bone formation and remodeling, resulting in pain from weak, malformed bone. |
18q21-qa22 |
LOH18CR1 |
IL-1/TNF signaling; RANKL signaling pathway |
| 5q31 |
| 5q35-qter |
| Retinoblastoma[22] |
Malignant tumor of the retina. Approximately 66% of patients are diagnosed by age 2 years and 95% of patients by age 3 years. Patients with heritable germ cell variants at greater risk of subsequent neoplasms. |
13q14.2 |
RB1 |
Cell-cycle checkpoint |
| Rothmund-Thomson syndrome (also called poikiloderma congenitale)[23,24] |
Autosomal recessive condition. Associated with skin findings (atrophy, telangiectasias, pigmentation), sparse hair, cataracts, small stature, and skeletal abnormalities. Increased incidence of osteosarcoma at a younger age. |
8q24.3 |
RECQL4 |
DNA helicase |
| Werner syndrome[25] |
Patients often have short stature and in their early twenties, develop signs of aging, including graying of hair and hardening of skin. Other aging problems such as cataracts, skin ulcers, and atherosclerosis develop later. |
8p12-p11.2 |
WRN |
DNA helicase; exonuclease activity |
For more information about these genetic syndromes, see the following summaries:
- Genetics of Breast and Gynecologic Cancers (Li-Fraumeni syndrome [LFS]).
- Genetics of Skin Cancer (Bloom syndrome, Rothmund-Thomson syndrome, and Werner syndrome).
References:
-
Chen X, Bahrami A, Pappo A, et al.: Recurrent somatic structural variations contribute to tumorigenesis in pediatric osteosarcoma. Cell Rep 7 (1): 104-12, 2014.
-
Perry JA, Kiezun A, Tonzi P, et al.: Complementary genomic approaches highlight the PI3K/mTOR pathway as a common vulnerability in osteosarcoma. Proc Natl Acad Sci U S A 111 (51): E5564-73, 2014.
-
Marinoff AE, Spurr LF, Fong C, et al.: Clinical Targeted Next-Generation Panel Sequencing Reveals MYC Amplification Is a Poor Prognostic Factor in Osteosarcoma. JCO Precis Oncol 7: e2200334, 2023.
-
Suehara Y, Alex D, Bowman A, et al.: Clinical Genomic Sequencing of Pediatric and Adult Osteosarcoma Reveals Distinct Molecular Subsets with Potentially Targetable Alterations. Clin Cancer Res 25 (21): 6346-6356, 2019.
-
Nacev BA, Sanchez-Vega F, Smith SA, et al.: Clinical sequencing of soft tissue and bone sarcomas delineates diverse genomic landscapes and potential therapeutic targets. Nat Commun 13 (1): 3405, 2022.
-
De Noon S, Ijaz J, Coorens TH, et al.: MYC amplifications are common events in childhood osteosarcoma. J Pathol Clin Res 7 (5): 425-431, 2021.
-
Parsons DW, Janeway KA, Patton DR, et al.: Actionable Tumor Alterations and Treatment Protocol Enrollment of Pediatric and Young Adult Patients With Refractory Cancers in the National Cancer Institute-Children's Oncology Group Pediatric MATCH Trial. J Clin Oncol 40 (20): 2224-2234, 2022.
-
Sanders RP, Drissi R, Billups CA, et al.: Telomerase expression predicts unfavorable outcome in osteosarcoma. J Clin Oncol 22 (18): 3790-7, 2004.
-
de Nonneville A, Salas S, Bertucci F, et al.: TOP3A amplification and ATRX inactivation are mutually exclusive events in pediatric osteosarcomas using ALT. EMBO Mol Med 14 (10): e15859, 2022.
-
Mirabello L, Zhu B, Koster R, et al.: Frequency of Pathogenic Germline Variants in Cancer-Susceptibility Genes in Patients With Osteosarcoma. JAMA Oncol 6 (5): 724-734, 2020.
-
Ognjanovic S, Olivier M, Bergemann TL, et al.: Sarcomas in TP53 germline mutation carriers: a review of the IARC TP53 database. Cancer 118 (5): 1387-96, 2012.
-
Mirabello L, Yeager M, Mai PL, et al.: Germline TP53 variants and susceptibility to osteosarcoma. J Natl Cancer Inst 107 (7): , 2015.
-
Toguchida J, Yamaguchi T, Dayton SH, et al.: Prevalence and spectrum of germline mutations of the p53 gene among patients with sarcoma. N Engl J Med 326 (20): 1301-8, 1992.
-
McIntyre JF, Smith-Sorensen B, Friend SH, et al.: Germline mutations of the p53 tumor suppressor gene in children with osteosarcoma. J Clin Oncol 12 (5): 925-30, 1994.
-
Maciaszek JL, Oak N, Chen W, et al.: Enrichment of heterozygous germline RECQL4 loss-of-function variants in pediatric osteosarcoma. Cold Spring Harb Mol Case Stud 5 (5): , 2019.
-
Kansara M, Thomas DM: Molecular pathogenesis of osteosarcoma. DNA Cell Biol 26 (1): 1-18, 2007.
-
German J: Bloom's syndrome. XX. The first 100 cancers. Cancer Genet Cytogenet 93 (1): 100-6, 1997.
-
Lipton JM, Federman N, Khabbaze Y, et al.: Osteogenic sarcoma associated with Diamond-Blackfan anemia: a report from the Diamond-Blackfan Anemia Registry. J Pediatr Hematol Oncol 23 (1): 39-44, 2001.
-
Idol RA, Robledo S, Du HY, et al.: Cells depleted for RPS19, a protein associated with Diamond Blackfan Anemia, show defects in 18S ribosomal RNA synthesis and small ribosomal subunit production. Blood Cells Mol Dis 39 (1): 35-43, 2007 Jul-Aug.
-
Li FP, Fraumeni JF, Mulvihill JJ, et al.: A cancer family syndrome in twenty-four kindreds. Cancer Res 48 (18): 5358-62, 1988.
-
Grimer RJ, Cannon SR, Taminiau AM, et al.: Osteosarcoma over the age of forty. Eur J Cancer 39 (2): 157-63, 2003.
-
Wong FL, Boice JD, Abramson DH, et al.: Cancer incidence after retinoblastoma. Radiation dose and sarcoma risk. JAMA 278 (15): 1262-7, 1997.
-
Wang LL, Gannavarapu A, Kozinetz CA, et al.: Association between osteosarcoma and deleterious mutations in the RECQL4 gene in Rothmund-Thomson syndrome. J Natl Cancer Inst 95 (9): 669-74, 2003.
-
Hicks MJ, Roth JR, Kozinetz CA, et al.: Clinicopathologic features of osteosarcoma in patients with Rothmund-Thomson syndrome. J Clin Oncol 25 (4): 370-5, 2007.
-
Goto M, Miller RW, Ishikawa Y, et al.: Excess of rare cancers in Werner syndrome (adult progeria). Cancer Epidemiol Biomarkers Prev 5 (4): 239-46, 1996.
Cellular Classification of Osteosarcoma and UPS of Bone
Osteosarcoma is a malignant tumor that is characterized by the direct formation of bone or osteoid tissue by the tumor cells. The World Health Organization's histological classification [1] of bone tumors separates the osteosarcomas into central (medullary) and surface (peripheral) tumors [2,3] and recognizes a number of subtypes within each group.
Central (Medullary) Tumors
- Conventional central osteosarcomas. The most common pathological subtype is conventional central osteosarcoma, which is characterized by areas of necrosis, atypical mitoses, and malignant osteoid tissue and/or cartilage. The other subtypes are much less common, each occurring at a frequency of less than 5%.
- Telangiectatic osteosarcomas.[4,5] Telangiectatic osteosarcoma may be confused radiographically with an aneurysmal bone cyst or giant cell tumor. This variant should be managed the same as a conventional osteosarcoma.[4,5]
- Intraosseous well-differentiated (low-grade) osteosarcomas.
- Small-cell osteosarcomas.
Surface (Peripheral) Tumors
The terms parosteal and periosteal osteosarcoma are embedded in the literature and widely used. They are confusing to patients and practitioners. It would be more helpful to divide osteosarcoma by location and histological grade. High-grade osteosarcoma, sometimes referred to as conventional osteosarcoma, typically arises centrally and grows outward, destroying surrounding cortex and soft tissues, but there are unequivocal cases of high-grade osteosarcoma in surface locations.[6] Similarly, there are reports of low-grade osteosarcoma arising in the medullary cavity.
- Parosteal (juxtacortical) well-differentiated (low-grade) osteosarcomas.[7,8] Parosteal osteosarcoma is defined as a lesion arising from the surface of the bone with a well-differentiated appearance on imaging and low-grade histological features.[9] The most common site for parosteal osteosarcoma is the posterior distal femur. Parosteal osteosarcoma occurs more often in older patients than does conventional high-grade osteosarcoma and is most common in patients aged 20 to 30 years. Parosteal osteosarcoma can be treated successfully with wide excision of the primary tumor alone.[7,10]
- Periosteal osteosarcomas (low-grade to intermediate-grade osteosarcomas).[11,12,13] Periosteal osteosarcoma typically appears as a broad-based soft tissue mass with extrinsic erosion of the underlying bony cortex.[12] Pathology shows an intermediate grade of differentiation. Wide resection is essential.
A single-institution retrospective review identified 29 patients with periosteal osteosarcoma.[11] The 5-year disease-free survival rate was 83%. The authors could not make a definitive statement regarding the benefits of adjuvant chemotherapy.
Another single-institution retrospective review identified 33 patients with periosteal osteosarcoma.[13] The 10-year overall survival (OS) rate was 84%. The 10-year OS rate was 83% for patients who were treated with surgery alone and 86% for patients who were treated with surgery and chemotherapy.
The European Musculoskeletal Oncology Society retrospectively analyzed 119 patients with periosteal osteosarcoma; 17 patients had metastasis.[12] The OS rate was 89% at 5 years and 83% at 10 years. Eighty-one patients received chemotherapy; 50 of those patients received chemotherapy before definitive surgical resection. There was no difference in outcome between the patients who received chemotherapy and the patients who did not receive chemotherapy.
- High-grade surface osteosarcomas.[3,6,14]
Extraosseous Osteosarcoma
Extraosseous osteosarcoma is a malignant mesenchymal neoplasm without direct attachment to the skeletal system. Previously, treatment for extraosseous osteosarcoma followed soft tissue sarcoma guidelines.[15] However, a retrospective analysis of the cooperative German-Austrian-Swiss osteosarcoma study group identified a favorable outcome for patients with extraosseous osteosarcoma who were treated with surgery and conventional osteosarcoma therapy.[16]
Undifferentiated Pleomorphic Sarcoma (UPS) of Bone
UPS of bone should be distinguished from angiomatoid fibrous histiocytoma, a low-grade tumor that is usually noninvasive, small, and associated with an excellent outcome using surgery alone.[17] One study suggests similar event-free survival rates for UPS and osteosarcoma.[18]
References:
-
Schajowicz F, Sissons HA, Sobin LH: The World Health Organization's histologic classification of bone tumors. A commentary on the second edition. Cancer 75 (5): 1208-14, 1995.
-
Antonescu CR, Huvos AG: Low-grade osteogenic sarcoma arising in medullary and surface osseous locations. Am J Clin Pathol 114 (Suppl): S90-103, 2000.
-
Kaste SC, Fuller CE, Saharia A, et al.: Pediatric surface osteosarcoma: clinical, pathologic, and radiologic features. Pediatr Blood Cancer 47 (2): 152-62, 2006.
-
Bacci G, Ferrari S, Ruggieri P, et al.: Telangiectatic osteosarcoma of the extremity: neoadjuvant chemotherapy in 24 cases. Acta Orthop Scand 72 (2): 167-72, 2001.
-
Weiss A, Khoury JD, Hoffer FA, et al.: Telangiectatic osteosarcoma: the St. Jude Children's Research Hospital's experience. Cancer 109 (8): 1627-37, 2007.
-
Okada K, Unni KK, Swee RG, et al.: High grade surface osteosarcoma: a clinicopathologic study of 46 cases. Cancer 85 (5): 1044-54, 1999.
-
Hoshi M, Matsumoto S, Manabe J, et al.: Oncologic outcome of parosteal osteosarcoma. Int J Clin Oncol 11 (2): 120-6, 2006.
-
Han I, Oh JH, Na YG, et al.: Clinical outcome of parosteal osteosarcoma. J Surg Oncol 97 (2): 146-9, 2008.
-
Kumar VS, Barwar N, Khan SA: Surface osteosarcomas: Diagnosis, treatment and outcome. Indian J Orthop 48 (3): 255-61, 2014.
-
Schwab JH, Antonescu CR, Athanasian EA, et al.: A comparison of intramedullary and juxtacortical low-grade osteogenic sarcoma. Clin Orthop Relat Res 466 (6): 1318-22, 2008.
-
Rose PS, Dickey ID, Wenger DE, et al.: Periosteal osteosarcoma: long-term outcome and risk of late recurrence. Clin Orthop Relat Res 453: 314-7, 2006.
-
Grimer RJ, Bielack S, Flege S, et al.: Periosteal osteosarcoma--a European review of outcome. Eur J Cancer 41 (18): 2806-11, 2005.
-
Cesari M, Alberghini M, Vanel D, et al.: Periosteal osteosarcoma: a single-institution experience. Cancer 117 (8): 1731-5, 2011.
-
Staals EL, Bacchini P, Bertoni F: High-grade surface osteosarcoma: a review of 25 cases from the Rizzoli Institute. Cancer 112 (7): 1592-9, 2008.
-
Wodowski K, Hill DA, Pappo AS, et al.: A chemosensitive pediatric extraosseous osteosarcoma: case report and review of the literature. J Pediatr Hematol Oncol 25 (1): 73-7, 2003.
-
Goldstein-Jackson SY, Gosheger G, Delling G, et al.: Extraskeletal osteosarcoma has a favourable prognosis when treated like conventional osteosarcoma. J Cancer Res Clin Oncol 131 (8): 520-6, 2005.
-
Daw NC, Billups CA, Pappo AS, et al.: Malignant fibrous histiocytoma and other fibrohistiocytic tumors in pediatric patients: the St. Jude Children's Research Hospital experience. Cancer 97 (11): 2839-47, 2003.
-
Picci P, Bacci G, Ferrari S, et al.: Neoadjuvant chemotherapy in malignant fibrous histiocytoma of bone and in osteosarcoma located in the extremities: analogies and differences between the two tumors. Ann Oncol 8 (11): 1107-15, 1997.
Staging and Site Information for Osteosarcoma and UPS of Bone
Historically, the Enneking staging system for skeletal malignancies was used to stage osteosarcoma and UPS of bone.[1] This system inferred the aggressiveness of the primary tumor by the descriptors intracompartmental or extracompartmental. The American Joint Committee on Cancer's tumor-node-metastasis (TNM) staging system for malignant bone tumors is not widely used for pediatric osteosarcoma, and patients are not stratified on the basis of prognostic stage groups.
For the purposes of treatment, osteosarcoma is described as one of the following:
- Localized. Patients without clinically detectable metastatic disease are considered to have localized osteosarcoma.
- Metastatic. Patients with any site of metastasis at the time of initial presentation detected by routine clinical studies are considered to have metastatic osteosarcoma.
Localized Osteosarcoma
Localized tumors are limited to the bone of origin. Patients with skip lesions confined to the bone that includes the primary tumor are considered to have localized disease if the skip lesions can be included in the planned surgical resection.[2] Approximately one-half of the tumors arise in the femur; of these, 80% are in the distal femur. Other primary sites, in descending order of frequency, are the proximal tibia, proximal humerus, pelvis, jaw, fibula, and ribs.[3] Osteosarcoma of the head and neck is more likely to be low grade [4] and to arise in older patients than is osteosarcoma of the appendicular skeleton.
Metastatic Osteosarcoma
Radiological evidence of metastatic tumor deposits is found in approximately 20% of patients at diagnosis, with 85% to 90% of metastatic disease presenting in the lungs. The second most common site of metastasis is another bone, which may be solitary or multiple.[5]
The syndrome of multifocal osteosarcoma refers to a presentation with multiple foci of osteosarcoma without a clear primary tumor, often with symmetrical metaphyseal involvement.[3]
Staging Evaluation
For patients with confirmed osteosarcoma, in addition to plain radiographs of the primary site that include a single-plane view of the entire affected bone to assess for skip metastasis, pretreatment staging studies should include the following:[6]
- Magnetic resonance imaging (MRI) of the primary site to include the entire bone.
- Computed tomography (CT) scan, if MRI is not available.
- Fluorine F 18-fludeoxyglucose (18F-FDG) positron emission tomography (PET)-CT or PET-MRI.[7,8]
- Bone scan if PET scan is not available.
- Posteroanterior and lateral chest radiograph.
- CT scan of the chest.
A retrospective review of 206 patients with osteosarcoma compared bone scan, PET scan, and PET-CT scan for the detection of bone metastases.[9] PET-CT was more sensitive and accurate than bone scan (sensitivity of 92% vs. 74%), and the combined use of both imaging studies achieved the highest sensitivity for diagnosing bone metastases in osteosarcoma (100%). 18F-FDG PET is the preferred staging modality for the detection of bone lesions. CT scan remains necessary for evaluation of pulmonary metastasis.
References:
-
Enneking WF: A system of staging musculoskeletal neoplasms. Clin Orthop Relat Res (204): 9-24, 1986.
-
Kager L, Zoubek A, Kastner U, et al.: Skip metastases in osteosarcoma: experience of the Cooperative Osteosarcoma Study Group. J Clin Oncol 24 (10): 1535-41, 2006.
-
Longhi A, Fabbri N, Donati D, et al.: Neoadjuvant chemotherapy for patients with synchronous multifocal osteosarcoma: results in eleven cases. J Chemother 13 (3): 324-30, 2001.
-
Patel SG, Meyers P, Huvos AG, et al.: Improved outcomes in patients with osteogenic sarcoma of the head and neck. Cancer 95 (7): 1495-503, 2002.
-
Harris MB, Gieser P, Goorin AM, et al.: Treatment of metastatic osteosarcoma at diagnosis: a Pediatric Oncology Group Study. J Clin Oncol 16 (11): 3641-8, 1998.
-
Meyer JS, Nadel HR, Marina N, et al.: Imaging guidelines for children with Ewing sarcoma and osteosarcoma: a report from the Children's Oncology Group Bone Tumor Committee. Pediatr Blood Cancer 51 (2): 163-70, 2008.
-
Oh C, Bishop MW, Cho SY, et al.: 18F-FDG PET/CT in the Management of Osteosarcoma. J Nucl Med 64 (6): 842-851, 2023.
-
Quartuccio N, Fox J, Kuk D, et al.: Pediatric bone sarcoma: diagnostic performance of ¹⁸F-FDG PET/CT versus conventional imaging for initial staging and follow-up. AJR Am J Roentgenol 204 (1): 153-60, 2015.
-
Byun BH, Kong CB, Lim I, et al.: Comparison of (18)F-FDG PET/CT and (99 m)Tc-MDP bone scintigraphy for detection of bone metastasis in osteosarcoma. Skeletal Radiol 42 (12): 1673-81, 2013.
Treatment Option Overview for Osteosarcoma and UPS of Bone
It is imperative that patients with proven or suspected osteosarcoma have an initial evaluation by an orthopedic oncologist familiar with the surgical management of this disease. This evaluation, which includes imaging studies, should be done before the initial biopsy because an inappropriately performed biopsy may jeopardize a limb-sparing procedure. Additionally, protective weight bearing is recommended for patients with tumors of weight-bearing bones to prevent pathological fractures that could preclude limb-preserving surgery.
Successful treatment generally requires the combination of effective systemic chemotherapy and complete resection of all clinically detectable disease.
Randomized clinical trials have established that both neoadjuvant and adjuvant chemotherapy are effective in preventing relapse in patients with clinically nonmetastatic tumors.[1]; [2][Level of evidence A1] The Pediatric Oncology Group (POG) conducted a study in which patients were randomly assigned to either immediate amputation or amputation after neoadjuvant therapy. A large percentage of patients declined to be assigned randomly, and the study was terminated without approaching the stated accrual goals. In the small number of patients treated, there was no difference in outcome between those who received preoperative chemotherapy and those who received postoperative chemotherapy.[3]
The treatment of osteosarcoma also depends on the histological grade, as follows:
- Low-grade osteosarcoma. Patients with low-grade osteosarcoma can be treated successfully by wide surgical resection alone, regardless of site of origin.
- Intermediate-grade osteosarcoma. Pathologists sometimes characterize tumors as intermediate-grade osteosarcoma. It is difficult to make treatment decisions for patients with intermediate-grade tumors. When a tumor biopsy suggests an intermediate-grade osteosarcoma, an option is to proceed with wide resection. The availability of the entire tumor allows the pathologist to examine more tissue and evaluate soft tissue and lymphovascular invasion, which can often clarify the nature of the lesion.
If the lesion proves to have high-grade elements, systemic chemotherapy is indicated, just as it would be for any high-grade osteosarcoma. The POG performed a study in which patients with high-grade osteosarcoma were randomly assigned to either immediate definitive surgery followed by adjuvant chemotherapy or to an initial period of chemotherapy followed by definitive surgery.[3] The outcome was the same for both groups. Although the strategy of initial chemotherapy followed by definitive surgery has become an almost universally applied approach for osteosarcoma, this study suggests that there is no increased risk of treatment failure if definitive surgery is done before chemotherapy begins; this can help to clarify equivocal diagnoses of intermediate-grade osteosarcoma.
- High-grade osteosarcoma. Patients with high-grade osteosarcoma require surgery and systemic chemotherapy. This treatment is necessary whether the tumor arises in the conventional central location or on a bone surface.
Recognition of intraosseous well-differentiated osteosarcoma and parosteal osteosarcoma is important because patients with these tumor types have the most favorable prognosis and can be treated successfully with wide excision of the primary tumor alone.[4,5] Patients with periosteal osteosarcoma have a generally good prognosis [6] and treatment is guided by histological grade.[5,7]
Patients with undifferentiated pleomorphic sarcoma (UPS) of bone are treated according to osteosarcoma treatment protocols.[8] A sarcoma-specific survival rate of 70.7% has been reported using primarily cisplatin- and doxorubicin-based regimens.[9]
Imaging modalities such as dynamic magnetic resonance imaging or positron emission tomography scanning are noninvasive methods to assess response,[10,11,12,13,14,15,16,17,18] and are the preferred modalities in the Children's Oncology Group AOST2032 (NCT05691478) trial.
Table 2 describes the treatment options for localized, metastatic, and recurrent osteosarcoma and UPS of bone.
Table 2. Treatment Options for Osteosarcoma and Undifferentiated Pleomorphic Sarcoma (UPS) of Bone
| Treatment Group |
Treatment Options |
| Localized osteosarcoma and UPS of bone |
Surgical removal of primary tumor. |
| Chemotherapy. |
| Radiation therapy, if surgery is not feasible or surgical margins are inadequate. |
| Osteosarcoma and UPS of bone with metastatic disease at diagnosis: |
Chemotherapy. |
| |
Lung-only metastases |
Preoperative chemotherapy followed by surgery to remove the tumor followed by postoperative combination chemotherapy. |
| |
Bone metastases with or without lung metastases |
Preoperative chemotherapy followed by surgery to remove the primary tumor and all metastatic disease followed by postoperative combination chemotherapy. |
| Surgery to remove the primary tumor followed by chemotherapy and then surgical resection of metastatic disease followed by postoperative combination chemotherapy. |
| Resection of metastatic bone lesions if possible. |
| Radiation therapy to the extremities (may offer some local control). |
| Recurrent osteosarcoma and UPS of bone: |
Surgery to remove all sites of metastatic disease. |
| Chemotherapy and targeted therapy. |
| Radiopharmaceuticals and radiation therapy. |
| |
Local recurrence |
Surgery to remove the tumor. |
| |
Lung-only recurrence |
Surgery to remove the tumor. |
| Chemotherapy or targeted therapy. |
| Radiation therapy. |
| |
Recurrence with bone-only metastases |
Surgery to remove the tumor. |
| 153Sm-EDTMP with or without stem cell support. |
| Chemotherapy or targeted therapy. |
| Radiation therapy. |
| |
Second recurrence of osteosarcoma |
Surgery to remove the tumor and/or chemotherapy. |
| Chemotherapy or targeted therapy. |
| 153Sm-EDTMP = samarium Sm 153-ethylenediamine tetramethylene phosphonic acid. |
References:
-
Link MP, Goorin AM, Miser AW, et al.: The effect of adjuvant chemotherapy on relapse-free survival in patients with osteosarcoma of the extremity. N Engl J Med 314 (25): 1600-6, 1986.
-
Bernthal NM, Federman N, Eilber FR, et al.: Long-term results (>25 years) of a randomized, prospective clinical trial evaluating chemotherapy in patients with high-grade, operable osteosarcoma. Cancer 118 (23): 5888-93, 2012.
-
Goorin AM, Schwartzentruber DJ, Devidas M, et al.: Presurgical chemotherapy compared with immediate surgery and adjuvant chemotherapy for nonmetastatic osteosarcoma: Pediatric Oncology Group Study POG-8651. J Clin Oncol 21 (8): 1574-80, 2003.
-
Hoshi M, Matsumoto S, Manabe J, et al.: Oncologic outcome of parosteal osteosarcoma. Int J Clin Oncol 11 (2): 120-6, 2006.
-
Schwab JH, Antonescu CR, Athanasian EA, et al.: A comparison of intramedullary and juxtacortical low-grade osteogenic sarcoma. Clin Orthop Relat Res 466 (6): 1318-22, 2008.
-
Rose PS, Dickey ID, Wenger DE, et al.: Periosteal osteosarcoma: long-term outcome and risk of late recurrence. Clin Orthop Relat Res 453: 314-7, 2006.
-
Grimer RJ, Bielack S, Flege S, et al.: Periosteal osteosarcoma--a European review of outcome. Eur J Cancer 41 (18): 2806-11, 2005.
-
Picci P, Bacci G, Ferrari S, et al.: Neoadjuvant chemotherapy in malignant fibrous histiocytoma of bone and in osteosarcoma located in the extremities: analogies and differences between the two tumors. Ann Oncol 8 (11): 1107-15, 1997.
-
Veitch ZW, Fasih S, Griffin AM, et al.: Clinical outcomes of non-osteogenic, non-Ewing soft-tissue sarcoma of bone--experience of the Toronto Sarcoma Program. Cancer Med 9 (24): 9282-9292, 2020.
-
Reddick WE, Wang S, Xiong X, et al.: Dynamic magnetic resonance imaging of regional contrast access as an additional prognostic factor in pediatric osteosarcoma. Cancer 91 (12): 2230-7, 2001.
-
Hawkins DS, Conrad EU, Butrynski JE, et al.: [F-18]-fluorodeoxy-D-glucose-positron emission tomography response is associated with outcome for extremity osteosarcoma in children and young adults. Cancer 115 (15): 3519-25, 2009.
-
Cheon GJ, Kim MS, Lee JA, et al.: Prediction model of chemotherapy response in osteosarcoma by 18F-FDG PET and MRI. J Nucl Med 50 (9): 1435-40, 2009.
-
Costelloe CM, Macapinlac HA, Madewell JE, et al.: 18F-FDG PET/CT as an indicator of progression-free and overall survival in osteosarcoma. J Nucl Med 50 (3): 340-7, 2009.
-
Hamada K, Tomita Y, Inoue A, et al.: Evaluation of chemotherapy response in osteosarcoma with FDG-PET. Ann Nucl Med 23 (1): 89-95, 2009.
-
Bajpai J, Kumar R, Sreenivas V, et al.: Prediction of chemotherapy response by PET-CT in osteosarcoma: correlation with histologic necrosis. J Pediatr Hematol Oncol 33 (7): e271-8, 2011.
-
Kong CB, Byun BH, Lim I, et al.: ¹⁸F-FDG PET SUVmax as an indicator of histopathologic response after neoadjuvant chemotherapy in extremity osteosarcoma. Eur J Nucl Med Mol Imaging 40 (5): 728-36, 2013.
-
Byun BH, Kong CB, Lim I, et al.: Combination of 18F-FDG PET/CT and diffusion-weighted MR imaging as a predictor of histologic response to neoadjuvant chemotherapy: preliminary results in osteosarcoma. J Nucl Med 54 (7): 1053-9, 2013.
-
Davis JC, Daw NC, Navid F, et al.: 18F-FDG Uptake During Early Adjuvant Chemotherapy Predicts Histologic Response in Pediatric and Young Adult Patients with Osteosarcoma. J Nucl Med 59 (1): 25-30, 2018.
Special Considerations for the Treatment of Children With Cancer
Cancer in children and adolescents is rare, although the overall incidence has been slowly increasing since 1975.[1] Children and adolescents with cancer should be referred to medical centers that have a multidisciplinary team of cancer specialists with experience treating the cancers that occur during childhood and adolescence. This multidisciplinary team approach incorporates the skills of the following pediatric specialists and others to ensure that children receive treatment, supportive care, and rehabilitation that will achieve optimal survival and quality of life:
- Primary care physicians.
- Pediatric surgeons.
- Transplant surgeons.
- Pathologists.
- Pediatric radiation oncologists.
- Pediatric medical oncologists and hematologists.
- Ophthalmologists.
- Rehabilitation specialists.
- Pediatric oncology nurses.
- Social workers.
- Child-life professionals.
- Psychologists.
- Nutritionists.
For specific information about supportive care for children and adolescents with cancer, see the summaries on Supportive and Palliative Care.
The American Academy of Pediatrics has outlined guidelines for pediatric cancer centers and their role in the treatment of children and adolescents with cancer.[2] At these centers, clinical trials are available for most types of cancer that occur in children and adolescents, and the opportunity to participate is offered to most patients and their families. Clinical trials for children and adolescents diagnosed with cancer are generally designed to compare potentially better therapy with current standard therapy. Other types of clinical trials test novel therapies when there is no standard therapy for a cancer diagnosis. Most of the progress in identifying curative therapies for childhood cancers has been achieved through clinical trials. Information about ongoing clinical trials is available from the NCI website.
Childhood and adolescent cancer survivors require close monitoring because side effects of cancer therapy may persist or develop months or years after treatment. For specific information about the incidence, type, and monitoring of late effects in childhood and adolescent cancer survivors, see Late Effects of Treatment for Childhood Cancer.
References:
-
Smith MA, Seibel NL, Altekruse SF, et al.: Outcomes for children and adolescents with cancer: challenges for the twenty-first century. J Clin Oncol 28 (15): 2625-34, 2010.
-
American Academy of Pediatrics: Standards for pediatric cancer centers. Pediatrics 134 (2): 410-4, 2014. Also available online. Last accessed December 15, 2023.
Treatment of Localized Osteosarcoma and UPS of Bone
Patients with localized osteosarcoma who undergo surgery and chemotherapy have a 5-year overall survival (OS) rate of 62% to 65%.[1] Complete surgical resection is crucial for patients with localized osteosarcoma, but it is not sufficient as the only therapy. At least 80% of patients treated with surgery alone will develop metastatic disease.[2] Randomized clinical trials have established that adjuvant chemotherapy is effective in preventing relapse or recurrence in patients with localized resectable primary tumors.[2]; [3][Level of evidence A1]
Undifferentiated pleomorphic sarcoma (UPS) of bone is seen more commonly in older adults. Patients with UPS of bone are treated according to osteosarcoma treatment protocols. The outcome for patients with resectable UPS is similar to the outcome for patients with osteosarcoma.[4] As with osteosarcoma, patients with favorable necrosis (≥90% necrosis) have a longer survival than do those with an inferior necrosis (<90% necrosis).[5] Many patients with UPS will need preoperative chemotherapy to achieve a wide local excision.[6]
Treatment Options for Localized Osteosarcoma and UPS of Bone
Treatment options for patients with localized osteosarcoma or UPS of bone include the following:
- Surgical removal of primary tumor.
- Chemotherapy (may start before or after definitive surgical resection of the primary tumor).
- Radiation therapy, if surgery is not feasible or surgical margins are inadequate.
Surgical removal of primary tumor
Surgical resection of the primary tumor with adequate margins is an essential component of the curative strategy for patients with localized osteosarcoma. The type of surgery required for complete ablation of the primary tumor depends on a number of factors that must be evaluated on a case-by-case basis.[7]
Limb-sparing procedures
In general, more than 80% of patients with extremity osteosarcoma can be treated using a limb-sparing procedure and do not require amputation.[8] Limb-sparing procedures are planned only when the preoperative staging indicates that it would be possible to achieve wide surgical margins. In one study, patients who underwent limb-salvage procedures who had poor histological response and close surgical margins had a high rate of local recurrence.[9]
Reconstruction after limb-sparing surgery can be accomplished with many options, including metallic endoprosthesis, allograft, vascularized autologous bone graft, and rotationplasty. An additional option, osteogenesis distraction bone transport, is available for patients whose tumors do not involve the epiphysis of long bones.[10] This procedure results in a stable reconstruction that functionally restores the normal limb.
The choice of optimal surgical reconstruction involves many factors, including the following:[11][Level of evidence A1]
- Site and size of the primary tumor.
- Ability to preserve the neurovascular supply of the distal extremity.
- Age of the patient and potential for additional growth.
- Needs and desires of the patient and family for specific functions such as sports participation.
If a complicated reconstruction delays or prohibits the resumption of systemic chemotherapy, limb preservation may endanger the chance for cure. In retrospective analyses of 703 patients with localized osteosarcoma, the resumption of chemotherapy 21 days or more after definitive surgery was associated with an increased risk of death and adverse events (hazard ratio [HR], 1.57; 1.04–2.36).[11] Delays in total time to completion of chemotherapy have also been associated with inferior OS and event-free survival (EFS). In a retrospective multivariate analysis of 113 patients with localized osteosarcoma, a delay of time to completion of chemotherapy greater than 4 weeks was associated with an OS HR of 2.70 (1.11–6.76, P = .003) and an EFS HR of 1.13 (1.00–1.26, P = .016).[12]
Amputation
For some patients, amputation remains the optimal choice for management of the primary tumor. A pathological fracture noted at diagnosis or during preoperative chemotherapy does not preclude limb-salvage surgery if wide surgical margins can be achieved.[13] If the pathological examination of the surgical specimen shows inadequate margins, an immediate amputation should be considered, especially if the histological necrosis after preoperative chemotherapy was poor.[14]
Factors associated with an increased risk of local recurrence
Patients who undergo amputation have lower local recurrence rates than do patients who undergo limb-salvage procedures.[15] However, there is no difference in OS between patients initially treated with amputation and those treated with a limb-sparing procedure. Patients with tumors of the femur have a higher local recurrence rate than do patients with primary tumors of the tibia or fibula. Rotationplasty and other limb-salvage procedures have been evaluated for both their functional outcome and their effect on survival. While limb-sparing resection is the current practice for local control at most pediatric institutions, there are few data to indicate that salvage of the lower limb is substantially superior to amputation with regard to patient quality of life.[16]
The German Cooperative Osteosarcoma Study Group performed a retrospective analysis of 1,802 patients with localized and metastatic osteosarcoma who underwent surgical resection of all clinically detectable disease.[17][Level of evidence C1] Local recurrence (n = 76) was associated with a high risk of death from osteosarcoma. Factors associated with an increased risk of local recurrence included nonparticipation in a clinical trial, pelvic primary site, limb-preserving surgery, soft tissue infiltration beyond the periosteum, poor pathological response to initial chemotherapy, failure to complete planned chemotherapy, and performing the biopsy at an institution different from where the definitive surgery is being performed.
Chemotherapy
Preoperative chemotherapy
Almost all patients receive intravenous preoperative chemotherapy as initial treatment. However, a standard chemotherapy regimen has not been determined. Current chemotherapy protocols include combinations of the following agents: high-dose methotrexate, doxorubicin, cyclophosphamide, cisplatin, ifosfamide, etoposide, and carboplatin.[18,19,20,21,22,23,24,25,26]
Evidence (preoperative chemotherapy):
- A meta-analysis of protocols for the treatment of osteosarcoma concluded the following:[27]
- Regimens containing three active chemotherapy agents were superior to regimens containing two active agents.
- Regimens with four active agents were not superior to regimens with three active agents.
- Three-drug regimens that did not include high-dose methotrexate were inferior to three-drug regimens that did include high-dose methotrexate.
- An Italian study that used regimens containing fewer courses of high-dose methotrexate observed the following results:[28][Level of evidence B4]
- There was a lower probability for EFS than found in earlier studies that used regimens containing more courses of high-dose methotrexate.
- The Children's Oncology Group (COG) performed a prospective randomized trial in newly diagnosed children and young adults with localized osteosarcoma. All patients received cisplatin, doxorubicin, and high-dose methotrexate. One-half of the patients were randomly assigned to receive ifosfamide. In a second randomization, one-half of the patients were assigned to receive the biological compound muramyl tripeptide-phosphatidyl ethanolamine encapsulated in liposomes (L-MTP-PE) beginning after definitive surgical resection. Results showed that:[29][Level of evidence A1]
- The addition of ifosfamide did not improve outcome.
- The addition of L-MTP-PE produced improvement in the EFS rate, which did not meet the conventional test for statistical significance (P = .08), and a significant improvement in the OS rate (78% vs. 70%; P = .03).
- There has been speculation regarding the potential contribution of postrelapse treatment, although there were no differences in the postrelapse surgical approaches in the relapsed patients. The appropriate role of L-MTP-PE in the treatment of osteosarcoma remains under discussion.[30]
- The COG performed a series of pilot studies in patients with newly diagnosed localized osteosarcoma.[31][Level of evidence B4]
- In pilot study 1, patients with lower degrees of necrosis after three-drug initial therapy received subsequent therapy with a higher cumulative dose of doxorubicin of 600 mg/m2.
- In pilot study 2, all patients received four-drug initial chemotherapy with cisplatin, doxorubicin, high-dose methotrexate, and ifosfamide. Patients with lower degrees of necrosis received subsequent chemotherapy with a higher cumulative dose of doxorubicin of 600 mg/m2.
- In pilot study 3, all patients received the same four-drug initial chemotherapy as pilot study 2. Patients with lower degrees of necrosis received higher doses of ifosfamide with the addition of etoposide in subsequent therapy.
The results of these pilot studies were as follows:
- Outcomes for all three pilot studies were similar to each other and to historical controls.
- All patients received dexrazoxane before each dose of doxorubicin. The addition of dexrazoxane did not appear to decrease the rate of good necrosis after initial therapy or EFS.
- Left ventricular fractional shortening, as measured by echocardiography, was minimally affected at 78 weeks from study entry.
- There was no evidence for an increased risk of secondary leukemia.
- The international European and American Osteosarcoma Study (EURAMOS) Group consortium was formed to conduct a large, prospective, randomized trial to help determine whether modifying the chemotherapy regimen on the basis of the degree of necrosis would improve EFS. All patients received initial therapy with cisplatin, doxorubicin, and high-dose methotrexate (MAP). Patients with more than 90% necrosis were randomly assigned to continue the same chemotherapy after surgery or to receive the same chemotherapy with the addition of interferon alpha-2B. Patients with less than 90% necrosis were randomly assigned to continue the same chemotherapy or to receive the same chemotherapy with the addition of high-dose ifosfamide and etoposide (MAPIE).[32][Level of evidence A1]
- At a median follow-up of 54 months for all registered patients (N = 2,260), the 3-year EFS rate was 59% (95% confidence interval [CI], 57%–61%), and the 5-year EFS rate was 54% (95% CI, 52%–56%).
- The 3-year OS rate was 79% (95% CI, 77%–81%), and the 5-year OS rate was 71% (95% CI, 68%–73%).
- Patients with localized disease at diagnosis (M0) who underwent complete surgical resection (n = 1,549) had 3-year EFS and OS rates from surgery of 70% and 88%, respectively; the 5-year EFS and OS rates from surgery were 64% and 79%, respectively.
- Forty percent of patients/families who participated in the study declined randomization after definitive surgical resection, making interpretation of the outcome of the randomized study questions more difficult and challenging the generalizability of the results.
- Of patients with more than 90% necrosis, 716 were randomly assigned to continue the same chemotherapy after surgery with or without the addition of interferon alpha-2B. The 3-year EFS rates were 74% (95% CI, 69%–79%) for patients who received MAP and 77% (95% CI, 72%–81%) for patients who received MAP plus interferon alpha-2B. The HR was 0.83 (95% CI, 0.61–1.12; P = .2).[33]
- Of patients with less than 90% necrosis, 618 were randomly assigned to continue MAP chemotherapy or to receive MAPIE chemotherapy. The 3-year EFS estimates for patients with localized disease were 60% (95% CI, 54%–66%) for the MAP group and 57% (95% CI, 51%–63%) for the MAPIE group. The HR was 1.03 (0.81–1.33, P = .80).[34]
Postoperative chemotherapy
Historically, the extent of tumor necrosis was used in some clinical trials to determine what type of postoperative chemotherapy would be given. In general, if tumor necrosis exceeded 90%, the preoperative chemotherapy regimen was continued. If tumor necrosis was less than 90%, some groups incorporated drugs not previously used in the preoperative therapy.
Patients with less necrosis after initial chemotherapy have an inferior prognosis than patients with more necrosis. The prognosis is still substantially better than the prognosis for patients treated with surgery alone and no adjuvant chemotherapy.
Based on the following evidence, it is inappropriate to conclude that patients with less necrosis have not responded to chemotherapy and that adjuvant chemotherapy should be withheld for these patients. Chemotherapy after definitive surgery should include the agents used in the initial phase of treatment unless there is clear and unequivocal progressive disease during the initial phase of therapy.
Evidence (using the same agents for postoperative chemotherapy):
- Early reports from the Memorial Sloan Kettering Cancer Center (MSKCC) suggested that adding cisplatin to postoperative chemotherapy improved the outcome for patients with less than 90% tumor necrosis.[35]
- With longer follow-up, the outcome for patients with less than 90% tumor necrosis treated at MSKCC was the same whether they did or did not receive cisplatin in the postoperative phase of treatment.[36]
- In an early experience, the German cooperative osteosarcoma group performed a trial in which the chemotherapy regimen for patients with poor necrosis was changed after initial treatment.[37] The agents used before surgery were discontinued and other agents were substituted.
- The results were substantially poorer for these patients than for patients who continued to receive the same agents.
- Subsequent trials performed by other groups failed to demonstrate improved EFS when drugs not included in the preoperative regimen (cisplatin) were added to postoperative therapy.[19,38]
- A limited-institution pilot trial tested the strategy of discontinuing the agents used in the initial phase of therapy for patients with poorer necrosis. Postoperative therapy consisted of melphalan with autologous stem cell reconstitution.[39]
- The 5-year EFS rate for this group was 28%, which was lower than the EFS rates observed in many large series in which agents were continued despite a lesser degree of necrosis.
- The international EURAMOS group consortium was formed to conduct a large, prospective, randomized trial to help determine whether modifying the chemotherapy regimen on the basis of the degree of necrosis would improve EFS. All patients received initial therapy with cisplatin, doxorubicin, and high-dose methotrexate (MAP). Patients with more than 90% necrosis were randomly assigned to continue the same chemotherapy after surgery or to receive the same chemotherapy with the addition of interferon alpha-2b.[33][Level of evidence B1] In the same EURAMOS trial, patients with less than 90% necrosis were randomly assigned to continue the same chemotherapy or to receive the same chemotherapy with the addition of high-dose ifosfamide and etoposide (MAPIE).[34][Level of evidence B1]
- The addition of interferon alpha-2b did not improve the probability of EFS.
- With a median follow-up of over 61 months, the EFS did not differ between the two groups.
- The intensification of treatment in the MAPIE group resulted in greater toxicity than did the treatment in the standard MAP arm.
Progression before local therapy
A single-institution retrospective analysis reported on early progression of osteosarcoma before local control.[40] Among 195 patients aged 18 years or younger, 25 (81%) had local-site progression only, and 6 patients had combined local- and metastatic-sites progression. The authors did not prospectively identify patients with clinical features that might suggest telangiectatic osteosarcoma with increased necrosis and hemorrhage, which might be an explanation for apparent progression. For the entire cohort, the 5-year EFS rate was 27.2%, and the OS rate was 31.3%. Patients with good necrosis had better 5-year EFS and OS rates (66.7% and 66.7%, respectively), compared with patients with a poor histological response (21.4% and 25.6%, respectively). However, these results did not reach statistical significance (P = .07 and P = .1).
Other chemotherapy approaches not considered effective
The Italian Sarcoma Group and the Scandinavian Sarcoma Group performed a clinical trial in patients with osteosarcoma who presented with clinically detectable metastatic disease.[41] Consolidation with high-dose etoposide and carboplatin followed by autologous stem cell reconstitution did not appear to improve outcome and the investigators did not recommend this strategy for the treatment of osteosarcoma.
Laboratory studies using cell lines and xenografts suggested that bisphosphonates had activity against osteosarcoma.[42] A single-institution clinical trial demonstrated that pamidronate could safely be administered along with multiagent chemotherapy to patients with newly diagnosed osteosarcoma.[42] The French pediatric and adult sarcoma cooperative groups performed a prospective trial for the treatment of osteosarcoma.[43] All patients received multiagent chemotherapy, and patients were randomly assigned to receive or not to receive zoledronate. The addition of zoledronate did not improve EFS.
Radiation therapy
If complete surgical resection is not feasible or if surgical margins are inadequate, radiation therapy may improve the local control rate.[44,45]; [46][Level of evidence C1] Radiation therapy should be considered in patients with osteosarcoma of the head and neck who have positive or uncertain resection margins.[47][Level of evidence C1]
Evidence (radiation therapy for local control):
- While it is accepted that the standard approach is primary surgical resection, a retrospective analysis of a small group of highly selective patients reported long-term EFS with external-beam radiation therapy for local control in some patients.[48][Level of evidence C1]
- Investigators from a single institution reported on 28 children and young adults with osteosarcoma who were treated with radiation therapy for local control. Sixteen patients received radiation therapy during the primary treatment course, and 12 patients received radiation therapy as part of therapy after recurrence.[49]
- For patients who received radiation therapy during primary treatment, the cumulative incidence of local failure at 5 years was 25%.
- For patients with recurrent disease, the cumulative incidence of local failure at 5 years was 44%.
- Local tumor progression was observed in 3 of 13 patients (23%) who were treated with adjuvant radiation therapy after resection, while three of six patients (50%) who received definitive radiation therapy as a sole modality of local control experienced local progression.
Osteosarcoma of the Head and Neck
Osteosarcoma of the head and neck occurs in an older population than does osteosarcoma of the extremities.[47,50,51,52,53] In the pediatric age group, osteosarcomas of the head and neck are more likely to be low-grade or intermediate-grade tumors than are osteosarcomas of the extremities.[54,55] All reported series emphasize the need for complete surgical resection.[47,50,51,52,53,54,55][Level of evidence C1] The probability for cure with surgery alone is higher for osteosarcoma of the head and neck than it is for extremity osteosarcoma. When surgical margins are positive, there is a trend for improved survival with adjuvant radiation therapy.[47,52][Level of evidence C1]
There are no randomized trials to assess the efficacy of chemotherapy in patients with osteosarcoma of the head and neck, but several series suggest a benefit.[50,56] Chemotherapy should be considered for younger patients with high-grade osteosarcoma of the head and neck.[54,57]
Patients with osteosarcoma of the head and neck have a higher risk of developing a local recurrence and a lower risk of having distant metastasis than do patients with osteosarcoma of the extremities.[50,52,53,58]
Current Clinical Trials
Use our advanced clinical trial search to find NCI-supported cancer clinical trials that are now enrolling patients. The search can be narrowed by location of the trial, type of treatment, name of the drug, and other criteria. General information about clinical trials is also available.
References:
-
Smith MA, Altekruse SF, Adamson PC, et al.: Declining childhood and adolescent cancer mortality. Cancer 120 (16): 2497-506, 2014.
-
Link MP, Goorin AM, Miser AW, et al.: The effect of adjuvant chemotherapy on relapse-free survival in patients with osteosarcoma of the extremity. N Engl J Med 314 (25): 1600-6, 1986.
-
Bernthal NM, Federman N, Eilber FR, et al.: Long-term results (>25 years) of a randomized, prospective clinical trial evaluating chemotherapy in patients with high-grade, operable osteosarcoma. Cancer 118 (23): 5888-93, 2012.
-
Picci P, Bacci G, Ferrari S, et al.: Neoadjuvant chemotherapy in malignant fibrous histiocytoma of bone and in osteosarcoma located in the extremities: analogies and differences between the two tumors. Ann Oncol 8 (11): 1107-15, 1997.
-
Bramwell VH, Steward WP, Nooij M, et al.: Neoadjuvant chemotherapy with doxorubicin and cisplatin in malignant fibrous histiocytoma of bone: A European Osteosarcoma Intergroup study. J Clin Oncol 17 (10): 3260-9, 1999.
-
Daw NC, Billups CA, Pappo AS, et al.: Malignant fibrous histiocytoma and other fibrohistiocytic tumors in pediatric patients: the St. Jude Children's Research Hospital experience. Cancer 97 (11): 2839-47, 2003.
-
Grimer RJ: Surgical options for children with osteosarcoma. Lancet Oncol 6 (2): 85-92, 2005.
-
Bacci G, Ferrari S, Bertoni F, et al.: Long-term outcome for patients with nonmetastatic osteosarcoma of the extremity treated at the istituto ortopedico rizzoli according to the istituto ortopedico rizzoli/osteosarcoma-2 protocol: an updated report. J Clin Oncol 18 (24): 4016-27, 2000.
-
Grimer RJ, Taminiau AM, Cannon SR, et al.: Surgical outcomes in osteosarcoma. J Bone Joint Surg Br 84 (3): 395-400, 2002.
-
Watanabe K, Tsuchiya H, Yamamoto N, et al.: Over 10-year follow-up of functional outcome in patients with bone tumors reconstructed using distraction osteogenesis. J Orthop Sci 18 (1): 101-9, 2013.
-
Imran H, Enders F, Krailo M, et al.: Effect of time to resumption of chemotherapy after definitive surgery on prognosis for non-metastatic osteosarcoma. J Bone Joint Surg Am 91 (3): 604-12, 2009.
-
Vasquez L, Silva J, Chavez S, et al.: Prognostic impact of diagnostic and treatment delays in children with osteosarcoma. Pediatr Blood Cancer 67 (4): e28180, 2020.
-
Scully SP, Ghert MA, Zurakowski D, et al.: Pathologic fracture in osteosarcoma : prognostic importance and treatment implications. J Bone Joint Surg Am 84-A (1): 49-57, 2002.
-
Bacci G, Ferrari S, Lari S, et al.: Osteosarcoma of the limb. Amputation or limb salvage in patients treated by neoadjuvant chemotherapy. J Bone Joint Surg Br 84 (1): 88-92, 2002.
-
Reddy KI, Wafa H, Gaston CL, et al.: Does amputation offer any survival benefit over limb salvage in osteosarcoma patients with poor chemonecrosis and close margins? Bone Joint J 97-B (1): 115-20, 2015.
-
Ottaviani G, Robert RS, Huh WW, et al.: Functional, psychosocial and professional outcomes in long-term survivors of lower-extremity osteosarcomas: amputation versus limb salvage. Cancer Treat Res 152: 421-36, 2009.
-
Andreou D, Bielack SS, Carrle D, et al.: The influence of tumor- and treatment-related factors on the development of local recurrence in osteosarcoma after adequate surgery. An analysis of 1355 patients treated on neoadjuvant Cooperative Osteosarcoma Study Group protocols. Ann Oncol 22 (5): 1228-35, 2011.
-
Fuchs N, Bielack SS, Epler D, et al.: Long-term results of the co-operative German-Austrian-Swiss osteosarcoma study group's protocol COSS-86 of intensive multidrug chemotherapy and surgery for osteosarcoma of the limbs. Ann Oncol 9 (8): 893-9, 1998.
-
Provisor AJ, Ettinger LJ, Nachman JB, et al.: Treatment of nonmetastatic osteosarcoma of the extremity with preoperative and postoperative chemotherapy: a report from the Children's Cancer Group. J Clin Oncol 15 (1): 76-84, 1997.
-
Bacci G, Picci P, Avella M, et al.: Effect of intra-arterial versus intravenous cisplatin in addition to systemic adriamycin and high-dose methotrexate on histologic tumor response of osteosarcoma of the extremities. J Chemother 4 (3): 189-95, 1992.
-
Cassano WF, Graham-Pole J, Dickson N: Etoposide, cyclophosphamide, cisplatin, and doxorubicin as neoadjuvant chemotherapy for osteosarcoma. Cancer 68 (9): 1899-902, 1991.
-
Voûte PA, Souhami RL, Nooij M, et al.: A phase II study of cisplatin, ifosfamide and doxorubicin in operable primary, axial skeletal and metastatic osteosarcoma. European Osteosarcoma Intergroup (EOI). Ann Oncol 10 (10): 1211-8, 1999.
-
Ferrari S, Smeland S, Mercuri M, et al.: Neoadjuvant chemotherapy with high-dose Ifosfamide, high-dose methotrexate, cisplatin, and doxorubicin for patients with localized osteosarcoma of the extremity: a joint study by the Italian and Scandinavian Sarcoma Groups. J Clin Oncol 23 (34): 8845-52, 2005.
-
Zalupski MM, Rankin C, Ryan JR, et al.: Adjuvant therapy of osteosarcoma--A Phase II trial: Southwest Oncology Group study 9139. Cancer 100 (4): 818-25, 2004.
-
Meyers PA, Schwartz CL, Krailo M, et al.: Osteosarcoma: a randomized, prospective trial of the addition of ifosfamide and/or muramyl tripeptide to cisplatin, doxorubicin, and high-dose methotrexate. J Clin Oncol 23 (9): 2004-11, 2005.
-
Daw NC, Neel MD, Rao BN, et al.: Frontline treatment of localized osteosarcoma without methotrexate: results of the St. Jude Children's Research Hospital OS99 trial. Cancer 117 (12): 2770-8, 2011.
-
Anninga JK, Gelderblom H, Fiocco M, et al.: Chemotherapeutic adjuvant treatment for osteosarcoma: where do we stand? Eur J Cancer 47 (16): 2431-45, 2011.
-
Ferrari S, Meazza C, Palmerini E, et al.: Nonmetastatic osteosarcoma of the extremity. Neoadjuvant chemotherapy with methotrexate, cisplatin, doxorubicin and ifosfamide. An Italian Sarcoma Group study (ISG/OS-Oss). Tumori 100 (6): 612-9, 2014 Nov-Dec.
-
Meyers PA, Schwartz CL, Krailo MD, et al.: Osteosarcoma: the addition of muramyl tripeptide to chemotherapy improves overall survival--a report from the Children's Oncology Group. J Clin Oncol 26 (4): 633-8, 2008.
-
Anderson PM, Meyers P, Kleinerman E, et al.: Mifamurtide in metastatic and recurrent osteosarcoma: a patient access study with pharmacokinetic, pharmacodynamic, and safety assessments. Pediatr Blood Cancer 61 (2): 238-44, 2014.
-
Schwartz CL, Wexler LH, Krailo MD, et al.: Intensified Chemotherapy With Dexrazoxane Cardioprotection in Newly Diagnosed Nonmetastatic Osteosarcoma: A Report From the Children's Oncology Group. Pediatr Blood Cancer 63 (1): 54-61, 2016.
-
Smeland S, Bielack SS, Whelan J, et al.: Survival and prognosis with osteosarcoma: outcomes in more than 2000 patients in the EURAMOS-1 (European and American Osteosarcoma Study) cohort. Eur J Cancer 109: 36-50, 2019.
-
Bielack SS, Smeland S, Whelan JS, et al.: Methotrexate, Doxorubicin, and Cisplatin (MAP) Plus Maintenance Pegylated Interferon Alfa-2b Versus MAP Alone in Patients With Resectable High-Grade Osteosarcoma and Good Histologic Response to Preoperative MAP: First Results of the EURAMOS-1 Good Response Randomized Controlled Trial. J Clin Oncol 33 (20): 2279-87, 2015.
-
Marina NM, Smeland S, Bielack SS, et al.: Comparison of MAPIE versus MAP in patients with a poor response to preoperative chemotherapy for newly diagnosed high-grade osteosarcoma (EURAMOS-1): an open-label, international, randomised controlled trial. Lancet Oncol 17 (10): 1396-1408, 2016.
-
Rosen G, Caparros B, Huvos AG, et al.: Preoperative chemotherapy for osteogenic sarcoma: selection of postoperative adjuvant chemotherapy based on the response of the primary tumor to preoperative chemotherapy. Cancer 49 (6): 1221-30, 1982.
-
Meyers PA, Heller G, Healey J, et al.: Chemotherapy for nonmetastatic osteogenic sarcoma: the Memorial Sloan-Kettering experience. J Clin Oncol 10 (1): 5-15, 1992.
-
Winkler K, Beron G, Delling G, et al.: Neoadjuvant chemotherapy of osteosarcoma: results of a randomized cooperative trial (COSS-82) with salvage chemotherapy based on histological tumor response. J Clin Oncol 6 (2): 329-37, 1988.
-
Smeland S, Müller C, Alvegard TA, et al.: Scandinavian Sarcoma Group Osteosarcoma Study SSG VIII: prognostic factors for outcome and the role of replacement salvage chemotherapy for poor histological responders. Eur J Cancer 39 (4): 488-94, 2003.
-
Venkatramani R, Murray J, Helman L, et al.: Risk-Based Therapy for Localized Osteosarcoma. Pediatr Blood Cancer 63 (3): 412-7, 2016.
-
Halalsheh H, Amer S, Sultan I: Progression before local control in osteosarcoma: Outcome and prognosis-predictive factors. Pediatr Blood Cancer 70 (11): e30649, 2023.
-
Boye K, Del Prever AB, Eriksson M, et al.: High-dose chemotherapy with stem cell rescue in the primary treatment of metastatic and pelvic osteosarcoma: final results of the ISG/SSG II study. Pediatr Blood Cancer 61 (5): 840-5, 2014.
-
Meyers PA, Healey JH, Chou AJ, et al.: Addition of pamidronate to chemotherapy for the treatment of osteosarcoma. Cancer 117 (8): 1736-44, 2011.
-
Piperno-Neumann S, Le Deley MC, Rédini F, et al.: Zoledronate in combination with chemotherapy and surgery to treat osteosarcoma (OS2006): a randomised, multicentre, open-label, phase 3 trial. Lancet Oncol 17 (8): 1070-80, 2016.
-
Ozaki T, Flege S, Kevric M, et al.: Osteosarcoma of the pelvis: experience of the Cooperative Osteosarcoma Study Group. J Clin Oncol 21 (2): 334-41, 2003.
-
DeLaney TF, Park L, Goldberg SI, et al.: Radiotherapy for local control of osteosarcoma. Int J Radiat Oncol Biol Phys 61 (2): 492-8, 2005.
-
Ciernik IF, Niemierko A, Harmon DC, et al.: Proton-based radiotherapy for unresectable or incompletely resected osteosarcoma. Cancer 117 (19): 4522-30, 2011.
-
Guadagnolo BA, Zagars GK, Raymond AK, et al.: Osteosarcoma of the jaw/craniofacial region: outcomes after multimodality treatment. Cancer 115 (14): 3262-70, 2009.
-
Hundsdoerfer P, Albrecht M, Rühl U, et al.: Long-term outcome after polychemotherapy and intensive local radiation therapy of high-grade osteosarcoma. Eur J Cancer 45 (14): 2447-51, 2009.
-
Tinkle CL, Lu J, Han Y, et al.: Curative-intent radiotherapy for pediatric osteosarcoma: The St. Jude experience. Pediatr Blood Cancer 66 (8): e27763, 2019.
-
Canadian Society of Otolaryngology-Head and Neck Surgery Oncology Study Group: Osteogenic sarcoma of the mandible and maxilla: a Canadian review (1980-2000). J Otolaryngol 33 (3): 139-44, 2004.
-
Kassir RR, Rassekh CH, Kinsella JB, et al.: Osteosarcoma of the head and neck: meta-analysis of nonrandomized studies. Laryngoscope 107 (1): 56-61, 1997.
-
Laskar S, Basu A, Muckaden MA, et al.: Osteosarcoma of the head and neck region: lessons learned from a single-institution experience of 50 patients. Head Neck 30 (8): 1020-6, 2008.
-
Patel SG, Meyers P, Huvos AG, et al.: Improved outcomes in patients with osteogenic sarcoma of the head and neck. Cancer 95 (7): 1495-503, 2002.
-
Gadwal SR, Gannon FH, Fanburg-Smith JC, et al.: Primary osteosarcoma of the head and neck in pediatric patients: a clinicopathologic study of 22 cases with a review of the literature. Cancer 91 (3): 598-605, 2001.
-
Daw NC, Mahmoud HH, Meyer WH, et al.: Bone sarcomas of the head and neck in children: the St Jude Children's Research Hospital experience. Cancer 88 (9): 2172-80, 2000.
-
Smeele LE, Snow GB, van der Waal I: Osteosarcoma of the head and neck: meta-analysis of the nonrandomized studies. Laryngoscope 108 (6): 946, 1998.
-
Smeele LE, Kostense PJ, van der Waal I, et al.: Effect of chemotherapy on survival of craniofacial osteosarcoma: a systematic review of 201 patients. J Clin Oncol 15 (1): 363-7, 1997.
-
Jasnau S, Meyer U, Potratz J, et al.: Craniofacial osteosarcoma Experience of the cooperative German-Austrian-Swiss osteosarcoma study group. Oral Oncol 44 (3): 286-94, 2008.
Treatment of Osteosarcoma and UPS of Bone With Metastatic Disease at Diagnosis
Approximately 20% to 25% of patients with osteosarcoma present with clinically detectable metastatic disease. For patients with metastatic disease at initial presentation, roughly 20% will remain continuously free of disease, and roughly 30% will survive 5 years from diagnosis.[1]
The lungs are the most common site of initial metastatic disease.[2] Patients with metastases limited to the lungs have a better outcome than do patients with metastases to other sites or to the lungs combined with other sites.[1,3]
Treatment Options for Osteosarcoma and UPS of Bone With Metastatic Disease at Diagnosis
Treatment options for patients with osteosarcoma or undifferentiated pleomorphic sarcoma (UPS) of bone with metastatic disease at diagnosis include the following:
- Chemotherapy.
The chemotherapeutic agents used include high-dose methotrexate, doxorubicin, cisplatin, high-dose ifosfamide, etoposide, and, in some reports, carboplatin or cyclophosphamide.
Evidence (chemotherapy):
- In a trial that investigated high-dose ifosfamide (17.5 g per course) in combination with etoposide for patients with newly diagnosed metastatic osteosarcoma, the following was observed:[4]
- The combination produced a complete response in 10% of the patients and a partial response in 49% of the patients.
However, similar to localized disease, there is no evidence that the addition of ifosfamide or etoposide contributes to improved event-free survival (EFS) or overall survival (OS) in patients with metastatic disease.
- A study using a factorial design in patients with metastatic osteosarcoma (n = 91) evaluated the addition of either muramyl tripeptide or ifosfamide to a standard chemotherapy regimen that included cisplatin, high-dose methotrexate, and doxorubicin.[5]
- There was a nominal advantage for the addition of muramyl tripeptide (but not for ifosfamide) in terms of EFS and OS, but criteria for statistical significance were not met.
- In the international European and American Osteosarcoma Study (EURAMOS) group consortium, 362 of 2,186 patients (17%) presented with metastasis at diagnosis. Patients were randomly assigned to receive either treatment with cisplatin, doxorubicin, and high-dose methotrexate or cisplatin, doxorubicin, high-dose methotrexate, and ifosfamide.[6][Level of evidence A1]
- At a median follow-up of 47 months, the 3-year EFS rate was 32% (95% confidence interval [CI], 27%–37%), and the 5-year EFS rate was 28% (95% CI, 23%–33%).
- The 3-year OS rate was 56% (95% CI, 50%–61%), and the 5-year OS rate was 45% (95% CI, 39%–50%).
The treatment options for UPS of bone with metastasis at initial presentation are the same as the treatment for osteosarcoma with metastasis. Patients with unresectable or metastatic UPS have a very poor outcome.[7]
Treatment Options for Lung-Only Metastases at Diagnosis
Treatment options for patients with metastatic lung lesions at diagnosis include the following:
- Preoperative chemotherapy followed by surgery to remove the tumor followed by postoperative combination chemotherapy.
Patients with metastatic lung lesions as the sole site of metastatic disease should have the lung lesions resected if possible. Generally, this is performed after the administration of preoperative chemotherapy. After definitive surgical resection of the primary tumor, most clinicians resume systemic chemotherapy before initiating lung surgery to avoid longer delays in the resumption of chemotherapy. In approximately 10% of patients, all lung lesions disappear after preoperative chemotherapy.[3] Complete resection of pulmonary metastatic disease can be achieved in a high percentage of patients with residual lung nodules after preoperative chemotherapy. The long-term survival is poor for patients who do not undergo complete surgical resection of pulmonary metastatic disease.[8,9][Level of evidence B4]
For patients who present with primary osteosarcoma and metastases limited to the lungs and who achieve complete surgical remission, the 5-year EFS rate is approximately 20% to 25%. Multiple metastatic nodules confer a worse prognosis than do one or two nodules, and bilateral lung involvement is worse than unilateral.[1] Patients with peripheral lung lesions may have a better prognosis than patients with central lesions.[10] Patients with fewer than three nodules confined to one lung may achieve a 5-year EFS rate of approximately 40% to 50%.[1]
A multi-institutional retrospective analysis compared thoracotomy with thoracoscopy for resection of pulmonary metastases in patients with osteosarcoma.[11] The analysis included patients who had pulmonary metastases at diagnosis, patients with pulmonary relapse after initial management of localized disease, and patients with disease progression while on therapy. The authors recognized a significant selection bias for the patients chosen to undergo thoracoscopy. In a Cox regression analysis, controlling for other factors impacting outcome, there was a significantly increased risk of mortality (hazard ratio [HR], 2.11; 95% CI, 1.09–4.09; P = .027) but not pulmonary recurrence (HR, 0.96; 95% CI, 0.52–1.79; P = .90) with a thoracoscopic approach. In a subset analysis limited to patients with oligometastatic disease, thoracoscopy did not increase the risk of mortality (HR, 1.16; 95% CI, 0.64–2.11; P = .62). The ongoing randomized trial (AOST2031 [NCT05235165]) was designed to definitively address this question and the selection bias. This trial will compare the effect of thoracotomy with thoracoscopic surgery.
Treatment Options for Bone Metastases With or Without Lung Metastases
The second most common site of metastasis is another bone that is distant from the primary tumor. Patients with metastasis to other bones distant from the primary tumor experience EFS and OS rates of approximately 10%.[1] In a study of patients who presented with primary extremity tumors and synchronous metastasis to other bones, only 3 of 46 patients remained continuously disease-free 5 years later.[12] Patients with transarticular skip lesions have a poor prognosis.[13]
Multifocal osteosarcoma is different from osteosarcoma that presents with a clearly delineated primary lesion and limited bone metastasis. Multifocal osteosarcoma classically presents with symmetrical, metaphyseal lesions, and it may be difficult to determine the primary lesion. Patients with multifocal bone disease at presentation have an extremely poor prognosis, but treatment with systemic chemotherapy and aggressive surgical resection may significantly prolong life.[14,15]
Treatment options for patients with bone metastases with or without lung metastases include the following:
- Preoperative chemotherapy followed by surgery to remove the primary tumor. After definitive surgical resection of the primary tumor, most clinicians resume systemic chemotherapy.
The timing of surgery to remove metastatic tumors is not well defined. It is usually not attempted at the time of primary surgery because delays of more than 21 days until resumption of chemotherapy can increase the risk of adverse events and death.[16]
- Surgery to remove the primary tumor followed by chemotherapy and then surgical resection of metastatic disease followed by postoperative combination chemotherapy.
When the usual treatment course of preoperative chemotherapy followed by surgical ablation of the primary tumor and resection of all overt metastatic disease followed by postoperative combination chemotherapy cannot be used, an alternative treatment approach may be used. This alternative treatment approach begins with surgery for the primary tumor, followed by chemotherapy, and then surgical resection of metastatic disease. This approach may be appropriate in patients with intractable pain, pathological fracture, or uncontrolled infection of the tumor when initiation of chemotherapy could create risk of sepsis.[17]
- Resection of metastatic bone lesions if possible.
- Radiation therapy to the extremities.
There is evidence that radiation therapy to the extremities may offer some local control.[18]
Treatment Options Under Clinical Evaluation
Information about National Cancer Institute (NCI)–supported clinical trials can be found on the NCI website. For information about clinical trials sponsored by other organizations, see the ClinicalTrials.gov website.
The following are examples of national and/or institutional clinical trials that are currently being conducted:
- AOST2031 (NCT05235165) (Thoracotomy Versus Thoracoscopic Management of Pulmonary Metastases in Patients With Osteosarcoma): This phase III trial compares the effect of thoracotomy with thoracoscopic surgery (video-assisted thoracoscopic surgery) in treating patients with osteosarcoma that has spread to the lung (pulmonary metastases). This trial is being done to evaluate the two different surgery methods for these patients and to determine which procedure is better.
- AOST2032 (NCT05691478) (A Study to Test the Addition of the Drug Cabozantinib to Chemotherapy in Patients With Newly Diagnosed Osteosarcoma): This trial starts with a feasibility phase for patients with high-risk osteosarcoma who have a resectable primary tumor. The feasibility phase will be followed by a randomized study comparing chemotherapy using methotrexate, doxorubicin, and cisplatin (MAP) with MAP and cabozantinib for patients with newly diagnosed localized and metastatic osteosarcoma.
Current Clinical Trials
Use our advanced clinical trial search to find NCI-supported cancer clinical trials that are now enrolling patients. The search can be narrowed by location of the trial, type of treatment, name of the drug, and other criteria. General information about clinical trials is also available.
References:
-
Kager L, Zoubek A, Pötschger U, et al.: Primary metastatic osteosarcoma: presentation and outcome of patients treated on neoadjuvant Cooperative Osteosarcoma Study Group protocols. J Clin Oncol 21 (10): 2011-8, 2003.
-
Kempf-Bielack B, Bielack SS, Jürgens H, et al.: Osteosarcoma relapse after combined modality therapy: an analysis of unselected patients in the Cooperative Osteosarcoma Study Group (COSS). J Clin Oncol 23 (3): 559-68, 2005.
-
Bacci G, Rocca M, Salone M, et al.: High grade osteosarcoma of the extremities with lung metastases at presentation: treatment with neoadjuvant chemotherapy and simultaneous resection of primary and metastatic lesions. J Surg Oncol 98 (6): 415-20, 2008.
-
Goorin AM, Harris MB, Bernstein M, et al.: Phase II/III trial of etoposide and high-dose ifosfamide in newly diagnosed metastatic osteosarcoma: a pediatric oncology group trial. J Clin Oncol 20 (2): 426-33, 2002.
-
Chou AJ, Kleinerman ES, Krailo MD, et al.: Addition of muramyl tripeptide to chemotherapy for patients with newly diagnosed metastatic osteosarcoma: a report from the Children's Oncology Group. Cancer 115 (22): 5339-48, 2009.
-
Smeland S, Bielack SS, Whelan J, et al.: Survival and prognosis with osteosarcoma: outcomes in more than 2000 patients in the EURAMOS-1 (European and American Osteosarcoma Study) cohort. Eur J Cancer 109: 36-50, 2019.
-
Daw NC, Billups CA, Pappo AS, et al.: Malignant fibrous histiocytoma and other fibrohistiocytic tumors in pediatric patients: the St. Jude Children's Research Hospital experience. Cancer 97 (11): 2839-47, 2003.
-
Yamamoto Y, Kanzaki R, Kanou T, et al.: Long-term outcomes and prognostic factors of pulmonary metastasectomy for osteosarcoma and soft tissue sarcoma. Int J Clin Oncol 24 (7): 863-870, 2019.
-
Slade AD, Warneke CL, Hughes DP, et al.: Effect of concurrent metastatic disease on survival in children and adolescents undergoing lung resection for metastatic osteosarcoma. J Pediatr Surg 50 (1): 157-60; discussion 160, 2015.
-
Letourneau PA, Xiao L, Harting MT, et al.: Location of pulmonary metastasis in pediatric osteosarcoma is predictive of outcome. J Pediatr Surg 46 (7): 1333-7, 2011.
-
Lautz TB, Farooqui Z, Jenkins T, et al.: Thoracoscopy vs thoracotomy for the management of metastatic osteosarcoma: A Pediatric Surgical Oncology Research Collaborative Study. Int J Cancer 148 (5): 1164-1171, 2021.
-
Bacci G, Fabbri N, Balladelli A, et al.: Treatment and prognosis for synchronous multifocal osteosarcoma in 42 patients. J Bone Joint Surg Br 88 (8): 1071-5, 2006.
-
Kager L, Zoubek A, Kastner U, et al.: Skip metastases in osteosarcoma: experience of the Cooperative Osteosarcoma Study Group. J Clin Oncol 24 (10): 1535-41, 2006.
-
Harris MB, Gieser P, Goorin AM, et al.: Treatment of metastatic osteosarcoma at diagnosis: a Pediatric Oncology Group Study. J Clin Oncol 16 (11): 3641-8, 1998.
-
Longhi A, Fabbri N, Donati D, et al.: Neoadjuvant chemotherapy for patients with synchronous multifocal osteosarcoma: results in eleven cases. J Chemother 13 (3): 324-30, 2001.
-
Imran H, Enders F, Krailo M, et al.: Effect of time to resumption of chemotherapy after definitive surgery on prognosis for non-metastatic osteosarcoma. J Bone Joint Surg Am 91 (3): 604-12, 2009.
-
Marina NM, Smeland S, Bielack SS, et al.: Comparison of MAPIE versus MAP in patients with a poor response to preoperative chemotherapy for newly diagnosed high-grade osteosarcoma (EURAMOS-1): an open-label, international, randomised controlled trial. Lancet Oncol 17 (10): 1396-1408, 2016.
-
Tinkle CL, Lu J, Han Y, et al.: Curative-intent radiotherapy for pediatric osteosarcoma: The St. Jude experience. Pediatr Blood Cancer 66 (8): e27763, 2019.
Treatment of Recurrent Osteosarcoma and UPS of Bone
Approximately 50% of relapses in patients with recurrent osteosarcoma occur within 18 months of therapy termination, and only 5% of recurrences develop beyond 5 years.[1,2,3,4]
Prognostic Factors for Recurrence
Prognostic factors for recurrent osteosarcoma or undifferentiated pleomorphic sarcoma (UPS) of bone include the following:
- Time from diagnosis. In 564 patients who experienced a recurrence, patients whose disease recurred within 2 years of diagnosis had a worse prognosis than patients whose disease recurred after 2 years.[1] In another series of 431 patients, recurrences occurring less than 2 years from diagnosis were also associated with worse outcomes.[5]
- Age at initial diagnosis. Older age at initial study enrollment was associated with a worse prognosis after recurrence.[5]
- Presence of metastatic disease at diagnosis. The presence of metastatic disease at initial presentation was associated with a worse prognosis after recurrence.[5]
- Tumor response to preoperative chemotherapy. Patients with a good histological response to initial preoperative chemotherapy had a better overall survival (OS) after recurrence than did patients with a poor initial response.[1]
- Site of metastases. In two large series, the incidence of recurrence by site was as follows: lung only (65%–80%), bone only (8%–10%), local recurrence only (4%–7%), and combined relapse (10%–15%).[4,6] Abdominal metastases are rare but may occur as late as 4 years after diagnosis.[7] The Children's Oncology Group (COG) reported the outcomes of 431 young patients with recurrent osteosarcoma.[5][Level of evidence C3] Patients with recurrences in both lung and bone had worse outcomes than did patients with recurrences in lung only (P = .005).
- Surgical resectability. Patients with recurrent osteosarcoma should be assessed for surgical resectability because they may sometimes be cured with aggressive surgical resection with or without chemotherapy.[8,6,9,10,11,12]
Control of osteosarcoma after recurrence depends on complete surgical resection of all sites of clinically detectable metastatic disease. If surgical resection is not attempted or cannot be performed, progression and death are certain. The ability to achieve a complete resection of recurrent disease is the most important prognostic factor at first relapse, with a 5-year survival rate of 20% to 45% after complete resection of metastatic pulmonary tumors and a 20% survival rate after complete resection of metastases at other sites.[4,6,12,13]
Treatment Options for Recurrent Osteosarcoma and UPS of Bone
Treatment options for patients with recurrent osteosarcoma or UPS of bone include the following:
- Surgery to remove all sites of metastatic disease.
- Chemotherapy and targeted therapy.
- Radiopharmaceuticals and radiation therapy.
Surgery
Control of osteosarcoma after recurrence depends on complete surgical resection of all sites of clinically detectable metastatic disease.
A multi-institutional retrospective analysis compared thoracotomy with thoracoscopy for resection of pulmonary metastases in patients with osteosarcoma.[14] The analysis included patients who had pulmonary metastases at diagnosis, patients with pulmonary relapse after initial management of localized disease, and patients with disease progression while on therapy. The authors recognized a significant selection bias for the patients chosen to undergo thoracoscopy. In a Cox regression analysis, controlling for other factors impacting outcome, there was a significantly increased risk of mortality (hazard ratio [HR], 2.11; 95% confidence interval [CI], 1.09–4.09; P = .027) but not pulmonary recurrence (HR, 0.96; 95% CI, 0.52–1.79; P = .90) with a thoracoscopic approach. In a subset analysis limited to patients with oligometastatic disease, thoracoscopy did not increase the risk of mortality (HR, 1.16; 95% CI, 0.64–2.11; P = .62). The ongoing randomized trial (AOST2031 [NCT05235165]) was designed to definitively address this question and the selection bias. This trial will compare the effect of thoracotomy with thoracoscopic surgery.
Chemotherapy and targeted therapy
The role of systemic chemotherapy for the treatment of patients with recurrent osteosarcoma is not well defined. The selection of further systemic treatment depends on many factors, including the site of recurrence, the patient's previous primary treatment, and individual patient considerations.
Osteosarcoma frequently has a stromal matrix that may further mineralize with tumor necrosis, leaving behind a mass seen on imaging that may or may not have a reduced number of tumor cells within it. Thus, standard Response Evaluation Criteria in Solid Tumors (RECIST) criteria may not be appropriate for evaluation of response to drugs in patients with osteosarcoma. The COG, in an attempt to establish baseline event-free survival (EFS) rates in patients with relapsed osteosarcoma, analyzed the outcomes of these patients from seven single-arm phase II trials. The drugs tested in each trial were determined to be inactive on the basis of radiographic response rates.[15]
- The EFS rate for 96 patients with osteosarcoma and measurable disease was 12% at 4 months (95% CI, 6%–19%).
- There was no significant difference in EFS between the trials according to the number of previous treatment regimens or patient age, sex, and ethnicity.
One additional phase II trial with a different study design was reported. In this trial, patients with osteosarcoma and metastases to the lung underwent surgical resection of all lung nodules and then were treated with adjuvant inhaled granulocyte-macrophage colony-stimulating factor (GM-CSF).[15]
- The 12-month EFS rate for the 42 evaluable patients enrolled in this study was 20% (95% CI, 10%–34%).
The following chemotherapy and targeted therapy agents have been studied to treat recurrent osteosarcoma and UPS of bone:
- Ifosfamide alone with mesna uroprotection, or in combination with etoposide. Ifosfamide alone with mesna uroprotection, or in combination with etoposide, has been active in as many as one-third of patients with recurrent osteosarcoma who have not previously received this drug.[16,17,18,19,20]
- Gemcitabine and docetaxel. A nonrandomized comparison of two doses of gemcitabine, both given with docetaxel, suggested that a higher dose of gemcitabine (900 mg/m2) was associated with a better response rate and longer survival than was a lower dose of gemcitabine (675 mg/m2) for patients with recurrent or refractory osteosarcoma.[21][Level of evidence C1] The combination of gemcitabine (at a dose of 900 mg/m2) and docetaxel has also been reported to have activity in some studies that included patients with unresectable disease.[22,23,24]; [25][Level of evidence C3]
- Cyclophosphamide and etoposide. Cyclophosphamide and etoposide have been shown to have activity in recurrent osteosarcoma.[22]
- Sorafenib. The Italian Sarcoma Group reported rare objective responses and disease stabilization with sorafenib in patients with recurrent osteosarcoma.[26]
- Sorafenib and everolimus. The Italian Sarcoma Group also reported the outcome of patients with metastatic recurrent osteosarcoma treated with the combination of sorafenib and everolimus. They observed two partial responses and two minor responses in 38 patients; 17 of 38 patients were progression free at 6 months from study entry but toxicity was greater than with sorafenib monotherapy.[27][Level of evidence B4]
- Regorafenib. Two prospective, randomized, double-blind trials have evaluated the role of regorafenib in the treatment of metastatic recurrent osteosarcoma. Both studies used the approved treatment regimen of 160 mg by mouth daily for 21 days followed by 7 days without treatment. The trial conducted in France included patients who were randomly assigned 2:1 between regorafenib and placebo and allowed crossover for patients assigned to placebo.[28] Seventeen of 26 patients (65%; one-sided 95% CI, 47%) in the regorafenib group did not have disease progression at 8 weeks, compared with 0 of 12 patients in the placebo group. The Sarcoma Alliance for Research Collaboration (SARC) group randomly assigned adult patients 1:1 between regorafenib and placebo.[29] Median progression-free survival (PFS) was significantly improved with regorafenib versus placebo: 3.6 months (95% CI, 2.0–7.6 months) versus 1.7 months (95% CI, 1.2–1.8 months), respectively (HR, 0.42; 95% CI, 0.21–0.85; P = .017).
- Dinutuximab in combination with GM-CSF. The COG performed a phase II study of dinutuximab, an antidisialoganglioside antibody, in combination with GM-CSF for patients with recurrent osteosarcoma. Of 39 patients, 28 experienced an event, and only 28.2% (95% CI, 15%–44.9%) of patients were event-free at the 12-month benchmark. Dinutuximab did not demonstrate sufficient evidence of efficacy.[30]
- Denosumab. The COG performed a phase II study of denosumab, a fully human monoclonal antibody to the receptor activator of nuclear factor-kappa beta ligand (RANKL). Patients aged 11 to 49 years with recurrent osteosarcoma were eligible for the study. Patients with measurable disease were eligible for cohort 1, and patients who underwent complete surgical resection of all sites of recurrent disease were eligible for cohort 2. The event-free rate did not exceed the historical rate experienced in previous COG trials to meet the study defined efficacy criteria in either cohort.[31]
- Lenvatinib with ifosfamide and etoposide. A multi-institutional group performed a phase I/II study using a combination of lenvatinib, ifosfamide, and etoposide to treat patients with relapsed or refractory osteosarcoma.[32][Level of evidence B4] The study suggested phase II doses for each drug: 1) 14 mg/m2 per day of lenvatinib (with a daily dose cap of 24 mg), 2) 100 mg/m2 per day of etoposide, and 3) 3,000 mg/m2 per day of ifosfamide. These drugs were given to patients intravenously on days 1 to 3 of each 3-week cycle for five cycles at most. Thirty-five patients from the phase I (cohort 3A; n = 15) and phase II (cohort 3B; n = 20) trials were treated at the recommended phase II doses, and their results were pooled. Eighteen of 35 patients had PFS rates of 51% (95% CI, 34%–69%) after 4 months, per the binomial estimate.
- Cabozantinib. A phase II clinical trial of this VEGFR2 and MET inhibitor evaluated 43 patients aged 12 years and older with relapsed osteosarcoma, 39 of whom had pulmonary metastases. The 6-month PFS rate was 33%, and five patients (12%) had a partial response.[33]
- Apatinib. A phase II clinical trial of this VEGFR2 inhibitor enrolled 37 patients aged 16 years and older. Sixteen patients (43%) had a partial response, and the 4-month PFS rate was 57%.[34] In a retrospective review of 19 patients with osteosarcoma who were treated with apatinib, 3 patients had a partial response (16%).[35]
- Anlotinib. A retrospective analysis reported the efficacy of the multitargeted tyrosine kinase inhibitor anlotinib in patients with recurrent metastatic osteosarcoma.[36] The study included 15 patients who were treated in China between June 2018 and April 2020. The median PFS was 9.8 months (± 0.9 months). The 6-month PFS rate was 73%, and the 10-month PFS rate was 33%. The median OS was 11.4 months (± 0.6 months). No patients achieved complete responses.
- Immunotherapy. Osteosarcoma frequently expresses PD-1, but trials of PD-1 inhibitors have been disappointing.[37,38,39] In these three studies, objective responses were observed in 1 of 14, 0 of 10, and 1 of 19 patients. HER2-expressing chimeric antigen receptor (CAR) T-cell therapy is also being studied.[40]
- High-dose chemotherapy with autologous hematopoietic stem cell transplant (HSCT). A study analyzed the addition of high-dose chemotherapy with autologous HSCT to treat Korean patients with relapsed osteosarcoma who achieved a complete response to salvage therapy.[41] Among 25 patients who achieved a complete response with salvage therapy, 15 were assigned to receive high-dose chemotherapy with autologous HSCT by investigator choice. In a subgroup analysis of outcomes in patients who achieved a complete response, there were no significant differences in the 5-year OS rates between patients who did and did not receive high-dose chemotherapy with autologous HSCT (83.9% ± 0.1% for 13 of 15 patients vs. 80.0% ± 0.1% for 8 of 10 patients, respectively; P = .923).
Radiopharmaceuticals and radiation therapy
High-dose samarium Sm 153-ethylenediamine tetramethylene phosphonic acid (153Sm-EDTMP) coupled with peripheral blood stem cell support may provide significant pain palliation in patients with bone metastases.[42,43,44,45] Toxicity of 153Sm-EDTMP is primarily hematologic.[46][Level of evidence C2]
A single-institution retrospective review reported that high-dose fraction radiation therapy (2 Gy/fraction) was a useful form of palliation for patients with recurrent osteosarcoma.[47][Level of evidence C3] Thirty-two courses of palliative radiation therapy were given to 20 patients with symptomatic metastatic and/or locally recurrent primary disease. Twenty-four of the 32 courses (75%) were associated with symptom improvement. Higher doses of radiation therapy correlated with longer durations of symptom response.
Palliation of painful lesions in children with recurrent or progressive disease can be achieved using a short course (10 or fewer fractions) of radiation therapy. In a retrospective study of 213 children with various malignancies, who were treated with short-course radiation therapy, 85% of patients had complete or partial pain relief, with low levels of toxicity.[48]
Treatment Options for Local Recurrence
Treatment options for patients with osteosarcoma or UPS of bone that has recurred locally include the following:
- Surgery to remove the tumor.
The postrelapse outcome of patients who have a local recurrence is quite poor.[49,50,51] Survival of patients with local recurrences and either previous or concurrent systemic metastases is poor.[52]
Two retrospective, single-institution series reported a survival rate of 10% to 40% after local recurrence without associated systemic metastasis.[52,53,54,55]
A retrospective review from the Italian Sarcoma Group identified 62 patients (median age, 21 years) with local recurrences.[56] With a median follow-up of 43 months (range, 5–235 months), the 5-year post–local relapse survival rate was 37%, significantly better for patients with a longer local recurrence–free interval (≤24 months, 31% vs. >24 months, 61.5%; P = .03), absence of distant metastases (no distant metastases, 56% vs. distant metastases, 11.5%; P = .0001), and achievement of second complete remission (CR) by surgical resection (no second CR, 0% vs. second CR, 58.5%; P = .0001). No difference in post–local relapse survival was found according to age, and there was no benefit from chemotherapy administration.
The incidence of local relapse was higher in patients who had a poor pathological response to chemotherapy in the primary tumor and in patients with inadequate surgical margins.[49,54]
Treatment Options for Lung-Only Recurrence
Treatment options for patients with osteosarcoma and UPS of bone that has recurred in the lung only include the following:
- Surgery to remove the tumor.
- Chemotherapy or targeted therapy. For more information, see the Chemotherapy and targeted therapy section.
- Radiation therapy.
Repeated resections of pulmonary recurrences can lead to extended disease control and, possibly, cure for some patients.[13,57] The survival rate is less than 5% for patients with unresectable metastatic disease.[6,58] The 5-year EFS rate ranges from 20% to 45% for patients who have complete surgical resection of all pulmonary metastases.[4,12,13]; [59][Level of evidence C1]
Factors associated with a better outcome include the following:[4,6,60,61,62]
- Fewer pulmonary nodules.
- Unilateral pulmonary metastases.
- Longer intervals between primary tumor resection and metastases.
- Tumor location in the periphery of the lung.
Approximately 50% of patients with one isolated pulmonary lesion more than 1 year after diagnosis were long-term survivors after metastasectomy. Chemotherapy did not appear to offer an advantage.[63][Level of evidence C1]
Control of osteosarcoma requires surgical resection of all macroscopic tumors. However, recommendations are conflicting regarding the surgical approach to the treatment of pulmonary metastases in osteosarcoma. Several options are available to resect pulmonary nodules in a patient with osteosarcoma, including thoracoscopy and thoracotomy with palpation of the collapsed lung. When patients have nodules identified only in one lung, some surgeons advocate thoracoscopy, some advocate unilateral thoracotomy, and some advocate bilateral thoracotomy. Bilateral thoracotomy can be performed as a single surgical procedure with a median sternotomy or a clamshell approach, or by staged bilateral thoracotomies.
Evidence (surgical approach for lung-only recurrence of osteosarcoma and UPS of bone):
- The St. Jude Children's Research Hospital reported on 81 patients who had pulmonary nodules identified at initial presentation in only one lung by computed tomography (CT) scan.[64] They performed unilateral thoracotomy and did not explore the contralateral hemithorax. At the time of thoracotomy, 44 of 81 patients had a solitary nodule identified; 15 of 81 patients had two nodules identified; 16 of 81 patients had three to five nodules identified; and 6 of 81 patients had more than five nodules identified. Additional patients who were considered unresectable were not included in the analysis.
- Thirty-nine of 81 patients had subsequent pulmonary recurrence; for most patients, recurrence occurred within 6 months.
- Within the first 6 months, 9 of 81 patients had ipsilateral recurrence, and 10 of 81 patients had a contralateral recurrence. By 2 years after initial thoracotomy, 13 of 81 patients had ipsilateral recurrence; 19 of 81 patients had contralateral recurrence; and 2 of 81 patients had bilateral recurrence.
- OS was similar for patients with ipsilateral and bilateral recurrence.
- The Memorial Sloan Kettering Cancer Center reported on pulmonary metastatic disease recurrence after initial therapy for osteosarcoma. Fourteen patients had pulmonary nodules identified in only one lung by CT scan. Nine patients were identified less than 2 years from initial diagnosis (early metastases), and five patients were identified more than 2 years from initial diagnosis (late metastases).[61] Seven of nine patients with early metastases had staged contralateral thoracotomies, and six of seven had nodules removed from the contralateral lung, despite negative CT scan findings.
- The lack of a comparison group precludes evaluation of the impact of the contralateral thoracotomy on subsequent EFS or OS.
- The same group expanded their analysis to include a retrospective review of 161 thoracotomies performed in 88 patients with osteosarcoma metastatic to the lung.[65] In this expanded series, CT failed to identify one-third of pulmonary metastases confirmed by pathological examination.
The COG is conducting a randomized trial (AOST2031 [NCT05235165]) to compare the effect of thoracotomy with thoracoscopic surgery to remove lung metastases. For more information, see the Treatment Options Under Clinical Evaluation section.
External-beam radiation therapy can provide local control of recurrent unresectable disease, symptomatic, and/or metastatic disease. Radiation therapy techniques allow for the delivery of very conformal high doses, known as stereotactic ablative radiation therapy (SABR) or stereotactic body radiation therapy (SBRT). SBRT and SABR administer treatment with high conformality and precision over a short period of time, providing good palliation and local control.[66]
Treatment Options for Recurrence With Bone-Only Metastases
Treatment options for patients with osteosarcoma or UPS of bone that has recurred in the bone only include the following:
- Surgery to remove the tumor.
- 153Sm-EDTMP with or without stem cell support.
- Chemotherapy or targeted therapy. For more information, see the Chemotherapy and targeted therapy section.
- Radiation therapy.
Patients with osteosarcoma who develop bone metastases have a poor prognosis. In one large series, the 5-year EFS rate was 11%.[67] Patients with late solitary bone relapse have a 5-year EFS rate of approximately 30%.[67,68,69,70]
For patients with multiple unresectable bone lesions, 153Sm-EDTMP with or without stem cell support may produce stable disease and/or provide pain relief.[46]
External-beam radiation therapy can provide local control of recurrent unresectable disease, symptomatic, and/or metastatic disease. Radiation therapy techniques allow for the delivery of very conformal high doses, known as SABR or SBRT. SBRT and SABR administer treatment with high conformality and precision over a short period of time, providing good palliation and local control.[66]
Treatment Options for Second Recurrence of Osteosarcoma
Treatment options for patients with osteosarcoma or UPS of bone that has recurred twice include the following:
- Surgery to remove the tumor and/or chemotherapy.
- Chemotherapy or targeted therapy. For more information, see the Chemotherapy and targeted therapy section.
Evidence (surgery and/or chemotherapy):
- The cooperative German-Austrian-Swiss osteosarcoma study group reported on 249 patients who had a second recurrence of osteosarcoma. The main therapy was repeated surgical resection of recurrent disease.[71]
- Of these patients, 197 died and 37 were alive in CR (24 patients after a third complete response and 13 patients after a fourth or subsequent complete response).
- Fifteen patients who did not achieve surgical remission remained alive, but follow-up for these patients was extremely short.
- The Spanish Group for Research on Sarcoma reported the results of a phase II trial of patients with relapsed or refractory osteosarcoma who were treated with gemcitabine and sirolimus.[72][Level of evidence C3]
- The PFS rate at 4 months was 44%.
- After central radiological review of 33 assessable patients, 2 partial responses and 14 disease stabilizations (48.5%) were reported.
- The COG reported the outcomes of patients with recurrent osteosarcoma from seven phase II trials, all of which were assessed to have shown no treatment benefit.[15]
- The EFS rate for 96 patients with osteosarcoma and measurable disease was 12% at 4 months (95% CI, 6%–19%).
- There was no significant difference in EFS between the trials according to the number of previous treatment regimens or patient age, sex, and ethnicity.
- One additional phase II trial with a different study design was reported. In this trial, patients with osteosarcoma and metastases to the lung underwent surgical resection of all lung nodules and then were treated with adjuvant inhaled GM-CSF.[15]
- The 12-month EFS rate for the 42 evaluable patients enrolled in this study was 20% (95% CI, 10%–34%).
Treatment Options Under Clinical Evaluation
Information about National Cancer Institute (NCI)–supported clinical trials can be found on the NCI website. For information about clinical trials sponsored by other organizations, see the ClinicalTrials.gov website.
The following are examples of national and/or institutional clinical trials that are currently being conducted:
- GD2-CAR PERSIST (NCT04539366) (Testing a New Immune Cell Therapy, GD2-Targeted Modified T-cells (GD2CART), in Children, Adolescents, and Young Adults with Relapsed/Refractory Osteosarcoma and Neuroblastoma): This is a phase I trial to determine the side effects and best dose of GD2CART to be effective against GD2-positive tumor cells.
- AOST2031 (NCT05235165) (Thoracotomy Versus Thoracoscopic Management of Pulmonary Metastases in Patients With Osteosarcoma): This phase III trial compares the effect of thoracotomy with thoracoscopic surgery (video-assisted thoracoscopic surgery) in treating patients with osteosarcoma that has spread to the lung (pulmonary metastases). This trial is being done to evaluate the two different surgery methods for these patients and to determine which procedure is better.
- NCT03628209 (Nivolumab With or Without Azacitidine in Treating Patients With Recurrent Resectable Osteosarcoma): This phase I/II trial will evaluate the safety and efficacy of nivolumab, or nivolumab in combination with azacitidine, in participants with recurrent, resectable osteosarcoma.
- NCT03811886 (Natalizumab for the Treatment of Recurrent, Refractory, or Progressive Pulmonary Metastatic Osteosarcoma): This is a phase I/II trial evaluating the safety of and response to natalizumab for patients with lung metastases that have progressed, relapsed, or become refractory to systemic therapy.
Current Clinical Trials
Use our advanced clinical trial search to find NCI-supported cancer clinical trials that are now enrolling patients. The search can be narrowed by location of the trial, type of treatment, name of the drug, and other criteria. General information about clinical trials is also available.
References:
-
Gelderblom H, Jinks RC, Sydes M, et al.: Survival after recurrent osteosarcoma: data from 3 European Osteosarcoma Intergroup (EOI) randomized controlled trials. Eur J Cancer 47 (6): 895-902, 2011.
-
Hauben EI, Bielack S, Grimer R, et al.: Clinico-histologic parameters of osteosarcoma patients with late relapse. Eur J Cancer 42 (4): 460-6, 2006.
-
Ferrari S, Briccoli A, Mercuri M, et al.: Late relapse in osteosarcoma. J Pediatr Hematol Oncol 28 (7): 418-22, 2006.
-
Kempf-Bielack B, Bielack SS, Jürgens H, et al.: Osteosarcoma relapse after combined modality therapy: an analysis of unselected patients in the Cooperative Osteosarcoma Study Group (COSS). J Clin Oncol 23 (3): 559-68, 2005.
-
Spraker-Perlman HL, Barkauskas DA, Krailo MD, et al.: Factors influencing survival after recurrence in osteosarcoma: A report from the Children's Oncology Group. Pediatr Blood Cancer 66 (1): e27444, 2019.
-
Bacci G, Briccoli A, Longhi A, et al.: Treatment and outcome of recurrent osteosarcoma: experience at Rizzoli in 235 patients initially treated with neoadjuvant chemotherapy. Acta Oncol 44 (7): 748-55, 2005.
-
Rejin K, Aykan OA, Omer G, et al.: Intra-abdominal metastasis in osteosarcoma: survey and literature review. Pediatr Hematol Oncol 28 (7): 609-15, 2011.
-
Goorin AM, Shuster JJ, Baker A, et al.: Changing pattern of pulmonary metastases with adjuvant chemotherapy in patients with osteosarcoma: results from the multiinstitutional osteosarcoma study. J Clin Oncol 9 (4): 600-5, 1991.
-
Harting MT, Blakely ML: Management of osteosarcoma pulmonary metastases. Semin Pediatr Surg 15 (1): 25-9, 2006.
-
Pastorino U, Gasparini M, Tavecchio L, et al.: The contribution of salvage surgery to the management of childhood osteosarcoma. J Clin Oncol 9 (8): 1357-62, 1991.
-
Skinner KA, Eilber FR, Holmes EC, et al.: Surgical treatment and chemotherapy for pulmonary metastases from osteosarcoma. Arch Surg 127 (9): 1065-70; discussion 1070-1, 1992.
-
Chou AJ, Merola PR, Wexler LH, et al.: Treatment of osteosarcoma at first recurrence after contemporary therapy: the Memorial Sloan-Kettering Cancer Center experience. Cancer 104 (10): 2214-21, 2005.
-
Harting MT, Blakely ML, Jaffe N, et al.: Long-term survival after aggressive resection of pulmonary metastases among children and adolescents with osteosarcoma. J Pediatr Surg 41 (1): 194-9, 2006.
-
Lautz TB, Farooqui Z, Jenkins T, et al.: Thoracoscopy vs thoracotomy for the management of metastatic osteosarcoma: A Pediatric Surgical Oncology Research Collaborative Study. Int J Cancer 148 (5): 1164-1171, 2021.
-
Lagmay JP, Krailo MD, Dang H, et al.: Outcome of Patients With Recurrent Osteosarcoma Enrolled in Seven Phase II Trials Through Children's Cancer Group, Pediatric Oncology Group, and Children's Oncology Group: Learning From the Past to Move Forward. J Clin Oncol 34 (25): 3031-8, 2016.
-
Harris MB, Cantor AB, Goorin AM, et al.: Treatment of osteosarcoma with ifosfamide: comparison of response in pediatric patients with recurrent disease versus patients previously untreated: a Pediatric Oncology Group study. Med Pediatr Oncol 24 (2): 87-92, 1995.
-
Miser JS, Kinsella TJ, Triche TJ, et al.: Ifosfamide with mesna uroprotection and etoposide: an effective regimen in the treatment of recurrent sarcomas and other tumors of children and young adults. J Clin Oncol 5 (8): 1191-8, 1987.
-
Kung FH, Pratt CB, Vega RA, et al.: Ifosfamide/etoposide combination in the treatment of recurrent malignant solid tumors of childhood. A Pediatric Oncology Group Phase II study. Cancer 71 (5): 1898-903, 1993.
-
Berrak SG, Pearson M, Berberoğlu S, et al.: High-dose ifosfamide in relapsed pediatric osteosarcoma: therapeutic effects and renal toxicity. Pediatr Blood Cancer 44 (3): 215-9, 2005.
-
Palmerini E, Setola E, Grignani G, et al.: High Dose Ifosfamide in Relapsed and Unresectable High-Grade Osteosarcoma Patients: A Retrospective Series. Cells 9 (11): , 2020.
-
Lee JA, Jeon DG, Cho WH, et al.: Higher Gemcitabine Dose Was Associated With Better Outcome of Osteosarcoma Patients Receiving Gemcitabine-Docetaxel Chemotherapy. Pediatr Blood Cancer 63 (9): 1552-6, 2016.
-
Massimo B, Giovanni G, Stefano F, et al.: Phase 2 trial of two courses of cyclophosphamide and etoposide for relapsed high-risk osteosarcoma patients. Cancer 115 (13): 2980-7, 2009.
-
Navid F, Willert JR, McCarville MB, et al.: Combination of gemcitabine and docetaxel in the treatment of children and young adults with refractory bone sarcoma. Cancer 113 (2): 419-25, 2008.
-
Qi WX, He AN, Tang LN, et al.: Efficacy and safety of gemcitabine-docetaxel combination therapy for recurrent or refractory high-grade osteosarcoma in China: a retrospective study of 18 patients. Jpn J Clin Oncol 42 (5): 427-31, 2012.
-
Palmerini E, Jones RL, Marchesi E, et al.: Gemcitabine and docetaxel in relapsed and unresectable high-grade osteosarcoma and spindle cell sarcoma of bone. BMC Cancer 16: 280, 2016.
-
Grignani G, Palmerini E, Dileo P, et al.: A phase II trial of sorafenib in relapsed and unresectable high-grade osteosarcoma after failure of standard multimodal therapy: an Italian Sarcoma Group study. Ann Oncol 23 (2): 508-16, 2012.
-
Grignani G, Palmerini E, Ferraresi V, et al.: Sorafenib and everolimus for patients with unresectable high-grade osteosarcoma progressing after standard treatment: a non-randomised phase 2 clinical trial. Lancet Oncol 16 (1): 98-107, 2015.
-
Duffaud F, Mir O, Boudou-Rouquette P, et al.: Efficacy and safety of regorafenib in adult patients with metastatic osteosarcoma: a non-comparative, randomised, double-blind, placebo-controlled, phase 2 study. Lancet Oncol 20 (1): 120-133, 2019.
-
Davis LE, Bolejack V, Ryan CW, et al.: Randomized Double-Blind Phase II Study of Regorafenib in Patients With Metastatic Osteosarcoma. J Clin Oncol 37 (16): 1424-1431, 2019.
-
Hingorani P, Krailo M, Buxton A, et al.: Phase 2 study of anti-disialoganglioside antibody, dinutuximab, in combination with GM-CSF in patients with recurrent osteosarcoma: A report from the Children's Oncology Group. Eur J Cancer 172: 264-275, 2022.
-
Janeway KA, Chou AJ, Hingorani P, et al.: AOST1321, a phase 2 trial of RANKL antibody, denosumab, in 2 cohorts of patients with recurrent or refractory osteosarcoma: a report from the Children's Oncology Group. [Abstract] 2019 Connective Tissue Oncology Society (CTOS) Annual Meeting, November 13-16, 2019, Tokyo, Japan. A-3254462, 2019. Available online. Last accessed March 30, 2023.
-
Gaspar N, Venkatramani R, Hecker-Nolting S, et al.: Lenvatinib with etoposide plus ifosfamide in patients with refractory or relapsed osteosarcoma (ITCC-050): a multicentre, open-label, multicohort, phase 1/2 study. Lancet Oncol 22 (9): 1312-1321, 2021.
-
Italiano A, Mir O, Mathoulin-Pelissier S, et al.: Cabozantinib in patients with advanced Ewing sarcoma or osteosarcoma (CABONE): a multicentre, single-arm, phase 2 trial. Lancet Oncol 21 (3): 446-455, 2020.
-
Xie L, Xu J, Sun X, et al.: Apatinib for Advanced Osteosarcoma after Failure of Standard Multimodal Therapy: An Open Label Phase II Clinical Trial. Oncologist 24 (7): e542-e550, 2019.
-
Tian Z, Liu H, Zhang F, et al.: Retrospective review of the activity and safety of apatinib and anlotinib in patients with advanced osteosarcoma and soft tissue sarcoma. Invest New Drugs 38 (5): 1559-1569, 2020.
-
Li H, Li Y, Song L, et al.: Retrospective review of safety and efficacy of anlotinib in advanced osteosarcoma with metastases after failure of standard multimodal therapy. Asia Pac J Clin Oncol 19 (5): e314-e319, 2023.
-
Tawbi HA, Burgess M, Bolejack V, et al.: Pembrolizumab in advanced soft-tissue sarcoma and bone sarcoma (SARC028): a multicentre, two-cohort, single-arm, open-label, phase 2 trial. Lancet Oncol 18 (11): 1493-1501, 2017.
-
Le Cesne A, Marec-Berard P, Blay JY, et al.: Programmed cell death 1 (PD-1) targeting in patients with advanced osteosarcomas: results from the PEMBROSARC study. Eur J Cancer 119: 151-157, 2019.
-
Boye K, Longhi A, Guren T, et al.: Pembrolizumab in advanced osteosarcoma: results of a single-arm, open-label, phase 2 trial. Cancer Immunol Immunother 70 (9): 2617-2624, 2021.
-
Ahmed N, Brawley VS, Hegde M, et al.: Human Epidermal Growth Factor Receptor 2 (HER2) -Specific Chimeric Antigen Receptor-Modified T Cells for the Immunotherapy of HER2-Positive Sarcoma. J Clin Oncol 33 (15): 1688-96, 2015.
-
Kang SH, Kim W, Lee JS, et al.: High-dose chemotherapy followed by autologous stem cell transplantation in pediatric patients with relapsed osteosarcoma. Pediatr Blood Cancer 70 (4): e30233, 2023.
-
Anderson PM, Wiseman GA, Dispenzieri A, et al.: High-dose samarium-153 ethylene diamine tetramethylene phosphonate: low toxicity of skeletal irradiation in patients with osteosarcoma and bone metastases. J Clin Oncol 20 (1): 189-96, 2002.
-
Franzius C, Bielack S, Flege S, et al.: High-activity samarium-153-EDTMP therapy followed by autologous peripheral blood stem cell support in unresectable osteosarcoma. Nuklearmedizin 40 (6): 215-20, 2001.
-
Sauerbrey A, Bielack S, Kempf-Bielack B, et al.: High-dose chemotherapy (HDC) and autologous hematopoietic stem cell transplantation (ASCT) as salvage therapy for relapsed osteosarcoma. Bone Marrow Transplant 27 (9): 933-7, 2001.
-
Fagioli F, Aglietta M, Tienghi A, et al.: High-dose chemotherapy in the treatment of relapsed osteosarcoma: an Italian sarcoma group study. J Clin Oncol 20 (8): 2150-6, 2002.
-
Loeb DM, Garrett-Mayer E, Hobbs RF, et al.: Dose-finding study of 153Sm-EDTMP in patients with poor-prognosis osteosarcoma. Cancer 115 (11): 2514-22, 2009.
-
Chen EL, Yoo CH, Gutkin PM, et al.: Outcomes for pediatric patients with osteosarcoma treated with palliative radiotherapy. Pediatr Blood Cancer 67 (1): e27967, 2020.
-
Sudmeier LJ, Madden N, Zhang C, et al.: Palliative radiotherapy for children: Symptom response and treatment-associated toxicity according to radiation therapy dose and fractionation. Pediatr Blood Cancer 70 (4): e30195, 2023.
-
Weeden S, Grimer RJ, Cannon SR, et al.: The effect of local recurrence on survival in resected osteosarcoma. Eur J Cancer 37 (1): 39-46, 2001.
-
Bacci G, Ferrari S, Lari S, et al.: Osteosarcoma of the limb. Amputation or limb salvage in patients treated by neoadjuvant chemotherapy. J Bone Joint Surg Br 84 (1): 88-92, 2002.
-
Rodriguez-Galindo C, Shah N, McCarville MB, et al.: Outcome after local recurrence of osteosarcoma: the St. Jude Children's Research Hospital experience (1970-2000). Cancer 100 (9): 1928-35, 2004.
-
Bacci G, Longhi A, Cesari M, et al.: Influence of local recurrence on survival in patients with extremity osteosarcoma treated with neoadjuvant chemotherapy: the experience of a single institution with 44 patients. Cancer 106 (12): 2701-6, 2006.
-
Grimer RJ, Sommerville S, Warnock D, et al.: Management and outcome after local recurrence of osteosarcoma. Eur J Cancer 41 (4): 578-83, 2005.
-
Bacci G, Forni C, Longhi A, et al.: Local recurrence and local control of non-metastatic osteosarcoma of the extremities: a 27-year experience in a single institution. J Surg Oncol 96 (2): 118-23, 2007.
-
Nathan SS, Gorlick R, Bukata S, et al.: Treatment algorithm for locally recurrent osteosarcoma based on local disease-free interval and the presence of lung metastasis. Cancer 107 (7): 1607-16, 2006.
-
Palmerini E, Torricelli E, Cascinu S, et al.: Is there a role for chemotherapy after local relapse in high-grade osteosarcoma? Pediatr Blood Cancer 66 (8): e27792, 2019.
-
Briccoli A, Rocca M, Salone M, et al.: Resection of recurrent pulmonary metastases in patients with osteosarcoma. Cancer 104 (8): 1721-5, 2005.
-
Tabone MD, Kalifa C, Rodary C, et al.: Osteosarcoma recurrences in pediatric patients previously treated with intensive chemotherapy. J Clin Oncol 12 (12): 2614-20, 1994.
-
Briccoli A, Rocca M, Salone M, et al.: High grade osteosarcoma of the extremities metastatic to the lung: long-term results in 323 patients treated combining surgery and chemotherapy, 1985-2005. Surg Oncol 19 (4): 193-9, 2010.
-
Aljubran AH, Griffin A, Pintilie M, et al.: Osteosarcoma in adolescents and adults: survival analysis with and without lung metastases. Ann Oncol 20 (6): 1136-41, 2009.
-
Su WT, Chewning J, Abramson S, et al.: Surgical management and outcome of osteosarcoma patients with unilateral pulmonary metastases. J Pediatr Surg 39 (3): 418-23; discussion 418-23, 2004.
-
Letourneau PA, Xiao L, Harting MT, et al.: Location of pulmonary metastasis in pediatric osteosarcoma is predictive of outcome. J Pediatr Surg 46 (7): 1333-7, 2011.
-
Daw NC, Chou AJ, Jaffe N, et al.: Recurrent osteosarcoma with a single pulmonary metastasis: a multi-institutional review. Br J Cancer 112 (2): 278-82, 2015.
-
Karplus G, McCarville MB, Smeltzer MP, et al.: Should contralateral exploratory thoracotomy be advocated for children with osteosarcoma and early unilateral pulmonary metastases? J Pediatr Surg 44 (4): 665-71, 2009.
-
Heaton TE, Hammond WJ, Farber BA, et al.: A 20-year retrospective analysis of CT-based pre-operative identification of pulmonary metastases in patients with osteosarcoma: A single-center review. J Pediatr Surg 52 (1): 115-119, 2017.
-
Brown LC, Lester RA, Grams MP, et al.: Stereotactic body radiotherapy for metastatic and recurrent ewing sarcoma and osteosarcoma. Sarcoma 2014: 418270, 2014.
-
Bacci G, Longhi A, Bertoni F, et al.: Bone metastases in osteosarcoma patients treated with neoadjuvant or adjuvant chemotherapy: the Rizzoli experience in 52 patients. Acta Orthop 77 (6): 938-43, 2006.
-
Aung L, Gorlick R, Healey JH, et al.: Metachronous skeletal osteosarcoma in patients treated with adjuvant and neoadjuvant chemotherapy for nonmetastatic osteosarcoma. J Clin Oncol 21 (2): 342-8, 2003.
-
Jaffe N, Pearson P, Yasko AW, et al.: Single and multiple metachronous osteosarcoma tumors after therapy. Cancer 98 (11): 2457-66, 2003.
-
Franke M, Hardes J, Helmke K, et al.: Solitary skeletal osteosarcoma recurrence. Findings from the Cooperative Osteosarcoma Study Group. Pediatr Blood Cancer 56 (5): 771-6, 2011.
-
Bielack SS, Kempf-Bielack B, Branscheid D, et al.: Second and subsequent recurrences of osteosarcoma: presentation, treatment, and outcomes of 249 consecutive cooperative osteosarcoma study group patients. J Clin Oncol 27 (4): 557-65, 2009.
-
Martin-Broto J, Redondo A, Valverde C, et al.: Gemcitabine plus sirolimus for relapsed and progressing osteosarcoma patients after standard chemotherapy: a multicenter, single-arm phase II trial of Spanish Group for Research on Sarcoma (GEIS). Ann Oncol 28 (12): 2994-2999, 2017.
Latest Updates to This Summary (06 / 17 / 2024)
The PDQ cancer information summaries are reviewed regularly and updated as new information becomes available. This section describes the latest changes made to this summary as of the date above.
Treatment of Recurrent Osteosarcoma and Undifferentiated Pleomorphic Sarcoma (UPS) of Bone
Added anlotinib as a targeted therapy agent that has been studied to treat recurrent osteosarcoma and UPS of bone. Also added text to state that a retrospective analysis reported the efficacy of the multitargeted tyrosine kinase inhibitor anlotinib in patients with recurrent metastatic osteosarcoma (cited Li et al. as reference 36). The study included 15 patients who were treated in China between June 2018 and April 2020. The median progression-free survival (PFS) was 9.8 months. The 6-month PFS rate was 73%, and the 10-month PFS rate was 33%. The median overall survival was 11.4 months. No patients achieved complete responses.
Added text to state that in three studies of PD-1 inhibitors, objective responses were observed in 1 of 14, 0 of 10, and 1 of 19 patients.
This summary is written and maintained by the PDQ Pediatric Treatment Editorial Board, which is editorially independent of NCI. The summary reflects an independent review of the literature and does not represent a policy statement of NCI or NIH. More information about summary policies and the role of the PDQ Editorial Boards in maintaining the PDQ summaries can be found on the About This PDQ Summary and PDQ® Cancer Information for Health Professionals pages.
About This PDQ Summary
Purpose of This Summary
This PDQ cancer information summary for health professionals provides comprehensive, peer-reviewed, evidence-based information about the treatment of osteosarcoma and undifferentiated pleomorphic sarcoma of bone. It is intended as a resource to inform and assist clinicians in the care of their patients. It does not provide formal guidelines or recommendations for making health care decisions.
Reviewers and Updates
This summary is reviewed regularly and updated as necessary by the PDQ Pediatric Treatment Editorial Board, which is editorially independent of the National Cancer Institute (NCI). The summary reflects an independent review of the literature and does not represent a policy statement of NCI or the National Institutes of Health (NIH).
Board members review recently published articles each month to determine whether an article should:
- be discussed at a meeting,
- be cited with text, or
- replace or update an existing article that is already cited.
Changes to the summaries are made through a consensus process in which Board members evaluate the strength of the evidence in the published articles and determine how the article should be included in the summary.
The lead reviewers for Osteosarcoma and Undifferentiated Pleomorphic Sarcoma of Bone Treatment are:
- Holcombe Edwin Grier, MD
- Karen J. Marcus, MD, FACR (Dana-Farber Cancer Institute/Boston Children's Hospital)
- William H. Meyer, MD
- Paul A. Meyers, MD (Memorial Sloan-Kettering Cancer Center)
- Thomas A. Olson, MD (Aflac Cancer and Blood Disorders Center of Children's Healthcare of Atlanta - Egleston Campus)
- Nita Louise Seibel, MD (National Cancer Institute)
- Malcolm A. Smith, MD, PhD (National Cancer Institute)
Any comments or questions about the summary content should be submitted to Cancer.gov through the NCI website's Email Us. Do not contact the individual Board Members with questions or comments about the summaries. Board members will not respond to individual inquiries.
Levels of Evidence
Some of the reference citations in this summary are accompanied by a level-of-evidence designation. These designations are intended to help readers assess the strength of the evidence supporting the use of specific interventions or approaches. The PDQ Pediatric Treatment Editorial Board uses a formal evidence ranking system in developing its level-of-evidence designations.
Permission to Use This Summary
PDQ is a registered trademark. Although the content of PDQ documents can be used freely as text, it cannot be identified as an NCI PDQ cancer information summary unless it is presented in its entirety and is regularly updated. However, an author would be permitted to write a sentence such as "NCI's PDQ cancer information summary about breast cancer prevention states the risks succinctly: [include excerpt from the summary]."
The preferred citation for this PDQ summary is:
PDQ® Pediatric Treatment Editorial Board. PDQ Osteosarcoma and Undifferentiated Pleomorphic Sarcoma of Bone Treatment. Bethesda, MD: National Cancer Institute. Updated <MM/DD/YYYY>. Available at: https://www.cancer.gov/types/bone/hp/osteosarcoma-treatment-pdq. Accessed <MM/DD/YYYY>. [PMID: 26389179]
Images in this summary are used with permission of the author(s), artist, and/or publisher for use within the PDQ summaries only. Permission to use images outside the context of PDQ information must be obtained from the owner(s) and cannot be granted by the National Cancer Institute. Information about using the illustrations in this summary, along with many other cancer-related images, is available in Visuals Online, a collection of over 2,000 scientific images.
Disclaimer
Based on the strength of the available evidence, treatment options may be described as either "standard" or "under clinical evaluation." These classifications should not be used as a basis for insurance reimbursement determinations. More information on insurance coverage is available on Cancer.gov on the Managing Cancer Care page.
Contact Us
More information about contacting us or receiving help with the Cancer.gov website can be found on our Contact Us for Help page. Questions can also be submitted to Cancer.gov through the website's Email Us.
Last Revised: 2024-06-17