General Information About Acute Myeloid Leukemia (AML)
AML is also called acute myelogenous leukemia and acute nonlymphocytic leukemia.
Incidence and Mortality
Estimated new cases and deaths from AML in the United States in 2024:[1]
- New cases: 20,800.
- Deaths: 11,220.
Based on Surveillance, Epidemiology, and End Results (SEER) Program data from 2013 to 2019, 31.7% of patients with AML were alive 5 years after diagnosis.[2]
Anatomy
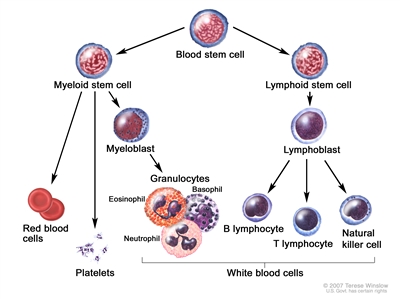
Blood cell development. A blood stem cell goes through several steps to become a red blood cell, platelet, or white blood cell.
AML is a heterogenous group of blood cancers that result from clonal expansion of myeloid hematopoietic precursors in the bone marrow. Not only are circulating leukemia cells (also called blasts) seen in the peripheral blood, but granulocytopenia, anemia, and thrombocytopenia are also common as proliferating leukemia cells interfere with normal hematopoiesis.[3]
Clinical Presentation
The diagnosis of AML is uncommon before age 45 years; the median age at diagnosis is 69 years.[2] Patients may present with symptoms that include the following:
- Weakness.
- Fever.
- Infection.
- Pallor.
- Bleeding.
The hampered production of normal blood cells due to leukemic infiltration of the bone marrow can also cause other symptoms and complications. Less commonly, patients have signs or symptoms related to the collection of leukemia cells in certain anatomical locations, such as central nervous system (CNS) or testicular involvement, or the presence of a myeloid sarcoma (also called chloroma). The symptoms of acute leukemia often arise over a 4- to 6-week period before diagnosis.[3]
Diagnostic Evaluation
The differentiation of AML from other forms of leukemia, in particular chronic myelogenous leukemia and acute lymphocytic leukemia, has vital therapeutic implications. The primary diagnostic tool in this determination is flow cytometry to evaluate surface antigens on the leukemia cells. Simple morphology is not adequate in determining lineage and, at a minimum, special histochemical stains are needed. While a diagnosis can be made by evaluating peripheral blood, a bone marrow biopsy is used to evaluate morphology and cell surface markers, as well as provide material for cytogenetic and molecular analysis. A peripheral blood or bone marrow blast count of 20% or greater is required to make the diagnosis, except for cases with certain chromosomal abnormalities (i.e., t(15;17), t(8;21), inv(16), or t(16;16)).[4]
Prognosis and Prognostic Factors
Advances in the treatment of AML have resulted in substantially improved complete remission (CR) rates.[2] Treatment should be sufficiently aggressive to achieve CR because partial remission offers no substantial survival benefit. Approximately 60% to 70% of adults with AML can be expected to attain CR status after appropriate induction therapy. More than 25% of adults with AML (about 45% of those who attain CR) can be expected to survive 3 or more years and may be cured.
Approximately half of patients with AML will harbor chromosomal abnormalities; therefore, conventional cytogenetic analysis remains mandatory in the evaluation of suspected AML.[5,6] With the routine use of molecular diagnostics, the identification of recurrent somatic mutations in NPM1, FLT3, CEPBA, and RUNX1, among other genes, has become a routine part of determining prognosis. Cytogenetic and molecular analyses provide the strongest prognostic information available, predicting outcome of both remission induction and postremission therapy.[7] Cytogenic and molecular information has been combined to form distinct prognostic groups.
Additional adverse prognostic factors for AML include the following:
- Age at diagnosis. Remission rates in adult AML are inversely related to age, with an expected remission rate of more than 65% for those younger than 60 years. Data suggest that once attained, duration of remission may be shorter in older patients. Increased morbidity and mortality during induction appear to be directly related to age.
- CNS involvement with leukemia.
- Systemic infection at diagnosis.
- Elevated white blood cell count (>100,000/mm3) at diagnosis.
- Therapy-related myeloid neoplasms, resulting from alkylating agents and radiation therapy.
- History of myelodysplastic syndrome or another antecedent hematologic disorder.
Long-Term Effects of Cancer Treatment
The risk of developing any long-term effects depends on the type and dose of treatment that was used and the age at which the patient underwent treatment.
A study of 30 patients who had AML that was in remission for at least 10 years demonstrated a 13% incidence of secondary malignancies.[8] Of 31 female long-term survivors of AML or acute lymphoblastic leukemia (ALL) diagnosed before age 40 years, 26 resumed normal menstruation after completion of therapy. Among 36 live offspring of survivors, two congenital problems occurred.[8]
Most patients with AML who undergo intensive therapy are treated with an anthracycline. Anthracyclines have been associated with increased risk of congestive heart failure (CHF).[9] Anthracycline cardiotoxicity is dose-dependent. In one study, doxorubicin-related CHF was 5% at a lifetime cumulative dose of 400 mg/m2, rising to 26% at a cumulative dose of 550 mg/m2.[10] In many cases, heart failure can manifest as a late effect.[11] In an analysis of children who underwent treatment for acute leukemia, the cumulative incidence of CHF at 10 years was 1.7% in ALL and 7.5% in AML.[12]
Patients who undergo allogeneic hematopoietic stem cell transplant can experience a large number of long-term or late side effects of treatment as a result of high-dose chemotherapy and/or radiation, and as an effect of chronic graft-versus-host disease and immunosuppression. These side effects may include chronic fatigue, thyroid and gonadal dysfunction, infertility, chronic infection, accelerated coronary heart disease, osteopenia, cataracts, iron overload, adverse psychological outcomes, and second cancers.[13,14,15]
In the Bone Marrow Transplant Survivor Study, hematopoietic cell transplant survivors had accelerated aging and were 8.4 times more likely to be frail than their siblings (95% confidence interval [CI], 2.0−34.5; P = .003). In a multivariable analysis, frailty was associated with a 2.76-fold increase in the risk of death, compared with a nonfrail state (95% CI, 1.7−4.4; P < .001).[16]
References:
-
American Cancer Society: Cancer Facts and Figures 2024. American Cancer Society, 2024. Available online. Last accessed June 21, 2024.
-
Surveillance, Epidemiology, and End Results Program: Cancer Stat Facts: Leukemia — Acute Myeloid Leukemia (AML). Bethesda, Md: National Cancer Institute, DCCPS, Surveillance Research Program, 2020. Available online. Last accessed October 5, 2023.
-
Sekeres MA, Gerds AT: Mitigating Fear and Loathing in Managing Acute Myeloid Leukemia. Semin Hematol 52 (3): 249-55, 2015.
-
Swerdlow SH, Campo E, Harris NL, et al., eds.: WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. 4th rev. ed. International Agency for Research on Cancer, 2017.
-
Slovak ML, Kopecky KJ, Cassileth PA, et al.: Karyotypic analysis predicts outcome of preremission and postremission therapy in adult acute myeloid leukemia: a Southwest Oncology Group/Eastern Cooperative Oncology Group Study. Blood 96 (13): 4075-83, 2000.
-
Grimwade D, Walker H, Harrison G, et al.: The predictive value of hierarchical cytogenetic classification in older adults with acute myeloid leukemia (AML): analysis of 1065 patients entered into the United Kingdom Medical Research Council AML11 trial. Blood 98 (5): 1312-20, 2001.
-
Döhner H, Estey E, Grimwade D, et al.: Diagnosis and management of AML in adults: 2017 ELN recommendations from an international expert panel. Blood 129 (4): 424-447, 2017.
-
Micallef IN, Rohatiner AZ, Carter M, et al.: Long-term outcome of patients surviving for more than ten years following treatment for acute leukaemia. Br J Haematol 113 (2): 443-5, 2001.
-
Steinherz LJ, Steinherz PG, Tan CT, et al.: Cardiac toxicity 4 to 20 years after completing anthracycline therapy. JAMA 266 (12): 1672-7, 1991.
-
Swain SM, Whaley FS, Ewer MS: Congestive heart failure in patients treated with doxorubicin: a retrospective analysis of three trials. Cancer 97 (11): 2869-79, 2003.
-
Hequet O, Le QH, Moullet I, et al.: Subclinical late cardiomyopathy after doxorubicin therapy for lymphoma in adults. J Clin Oncol 22 (10): 1864-71, 2004.
-
Chellapandian D, Pole JD, Nathan PC, et al.: Congestive heart failure among children with acute leukemia: a population-based matched cohort study. Leuk Lymphoma 60 (2): 385-394, 2019.
-
Inamoto Y, Lee SJ: Late effects of blood and marrow transplantation. Haematologica 102 (4): 614-625, 2017.
-
Sun CL, Francisco L, Baker KS, et al.: Adverse psychological outcomes in long-term survivors of hematopoietic cell transplantation: a report from the Bone Marrow Transplant Survivor Study (BMTSS). Blood 118 (17): 4723-31, 2011.
-
Armenian SH, Sun CL, Kawashima T, et al.: Long-term health-related outcomes in survivors of childhood cancer treated with HSCT versus conventional therapy: a report from the Bone Marrow Transplant Survivor Study (BMTSS) and Childhood Cancer Survivor Study (CCSS). Blood 118 (5): 1413-20, 2011.
-
Arora M, Sun CL, Ness KK, et al.: Physiologic Frailty in Nonelderly Hematopoietic Cell Transplantation Patients: Results From the Bone Marrow Transplant Survivor Study. JAMA Oncol 2 (10): 1277-1286, 2016.
Classification of AML
World Health Organization (WHO) Classification
The classification of acute myeloid leukemia (AML) has been revised by a group of pathologists and clinicians under the auspices of the WHO.[1] While elements of the French-American-British (FAB) classification have been retained (i.e., morphology, immunophenotype, cytogenetics, and clinical features),[2,3] the WHO classification incorporates and interrelates morphology, cytogenetics, molecular genetics, and immunologic markers, which construct a classification that is universally applicable and has prognostic and therapeutic relevance.[1,3,4] Each criterion has prognostic and treatment implications but, for practical purposes, initial antileukemic therapy is similar for all subtypes.
In 2001, the WHO proposed a new classification system that incorporated diagnostic cytogenetic information and that more reliably correlated with outcome. This classification system also decreased the bone marrow percentage of leukemic blast requirement for the diagnosis of AML from 30% to 20%. An additional clarification was made so patients with recurrent cytogenetic abnormalities did not need to meet the minimum blast requirement to be considered as having an AML diagnosis.[5,6,7]
In 2008, the WHO expanded the number of cytogenetic abnormalities linked to AML classification and, for the first time, included specific gene mutations (CEBPA and NPM) in its classification system.[5,8] With the addition of these gene mutations, FAB subclassification no longer provided prognostic information for patients with a diagnosis of AML, not otherwise specified (NOS).[9]
In 2016, the WHO classification underwent revisions to incorporate the expanding knowledge of leukemia biomarkers that are significantly important to the diagnosis, prognosis, and treatment of leukemia.[10] With emerging technologies aimed at genetic, epigenetic, proteomic, and immunophenotypic classification, AML classification will continue to evolve and provide informative prognostic and biological guidelines to clinicians and researchers.
2016 WHO classification of AML and related neoplasms
- AML with recurrent genetic abnormalities:
- AML with t(8;21)(q22;q22), RUNX1::RUNX1T1.
- AML with inv(16)(p13.1;q22) or t(16;16)(p13.1;q22), CBFB::MYH11.
- Acute promyelocytic leukemia (APL) with PML::RARA.
- AML with t(9;11)(p21.3;q23.3), MLLT3::KMT2A.
- AML with t(6;9)(p23;q34.1), DEK::NUP214.
- AML with inv(3)(q21.3;q26.2) or t(3;3)(q21.3;q26.2), GATA2, MECOM.
- AML (megakaryoblastic) with t(1;22)(p13.3;q13.3), RBM15::MKL1.
- AML with BCR::ABL1 (provisional entity).
- AML with mutated NPM1.
- AML with biallelic mutations of CEBPA.
- AML with mutated RUNX1 (provisional entity).
- AML with myelodysplasia-related features.
- Therapy-related myeloid neoplasms.
- AML, NOS:
- AML with minimal differentiation (FAB classification M0).
- AML without maturation (FAB classification M1).
- AML with maturation (FAB classification M2).
- Acute myelomonocytic leukemia (FAB classification M4).
- Acute monoblastic/monocytic leukemia (FAB classification M5a and M5b).
- Pure erythroid leukemia (FAB classification M6a and M6b).
- Acute megakaryoblastic leukemia (FAB classification M7).
- Acute basophilic leukemia.
- Acute panmyelosis with myelofibrosis.
- Myeloid sarcoma.
- Myeloid proliferations related to Down syndrome:
- Transient abnormal myelopoiesis (TAM).
- Myeloid leukemia associated with Down syndrome.
AML With Recurrent Genetic Abnormalities
AML with well-defined genetic abnormalities is characterized by recurrent genetic abnormalities.[10] The reciprocal translocations t(8;21), inv(16) or t(16;16), t(15;17), and translocations involving the 11q23 breakpoint are the most commonly identified chromosomal abnormalities. These structural chromosome rearrangements result in the formation of fusion genes that encode chimeric proteins that may contribute to the initiation or progression of leukemogenesis. Many of these translocations are detected by either reverse transcriptase–polymerase chain reaction (RT–PCR) or fluorescence in situ hybridization (FISH), which has a higher sensitivity than metaphase cytogenetics. Other recurring cytogenetic abnormalities are less common.
Molecular diagnostic platforms such as next-generation sequencing along with RT-PCR are used to identify recurrent molecular abnormalities in AML, helping to further refine diagnostic categories in the 2016 WHO classification system.[10]
AML with t(8;21)(q22;q22), RUNX1-RUNX1T1
The translocation t(8;21)(q22;q22) is one of the most common chromosomal aberrations in AML and accounts for 5% to 12% of cases.[11] Myeloid sarcomas (chloromas) may be present and may be associated with a bone marrow blast percentage of less than 20%.
Common morphological features include the following:
- Large blasts with abundant basophilic cytoplasm, often containing numerous azurophilic granules.
- A few blasts in some cases show very large granules (pseudo Chediak-Higashi granules).
- Auer rods, which may be detected in mature neutrophils.
- Smaller blasts, predominantly in the peripheral blood.
- Promyelocytes, myelocytes, and mature neutrophils with variable dysplasia in the bone marrow.
- Abnormal nuclear segmentation (pseudo Pelger-Huët nuclei) and/or cytoplasmic staining abnormalities.
- Increased eosinophil precursors.
- Reduced or absent monocytes.
- Normal erythroblasts and megakaryocytes.
Rarely, AML with this translocation presents with a bone marrow blast percentage of less than 20%.[5] Along with inv(16)(p13;q22) or t(16;16)(p13;q22), AML with t(8;21) makes up a category known as core binding factor AML. This category of AML is associated with long-term survival when treated with high-dose cytarabine.[12,13,14,15]
The translocation t(8;21)(q22;q22) involves the RUNX1 gene, which encodes CBF-alpha, and the RUNX1T1 (8;21) gene.[5,16] The RUNX1::RUNX1T1 fusion transcript is consistently detected in patients with t(8;21) AML. This translocation is usually associated with a good response to chemotherapy and a high complete remission (CR) rate with long-term survival when treated with high-dose cytarabine in the postremission phase, as demonstrated in the Cancer and Leukemia Group B (CLB-9022 and CLB-8525) trials.[12,13,14,15] Additional chromosome abnormalities are common, for example, loss of a sex chromosome and del(9)(q22). Leukocytosis (i.e., white blood count >25 × 109 /L) is associated with an inferior outcome,[17] as is the presence of a KIT mutation.[18]
AML with inv(16)(p13.1;q22) or t(16;16)(p13.1;q22), CBFB::MYH11
The inv(16)(p13;q22) abnormality or t(16;16)(p13;q22) translocation is found in approximately 10% to 12% of all cases of AML, predominantly in younger patients.[5,19] Myeloid sarcomas may be present at initial diagnosis or at relapse.
Common morphological features include the following:
- Monocytic and granulocytic differentiation.
- A characteristically abnormal eosinophil component with immature purple-violet eosinophil granules that may obscure cell morphology if present in great numbers.
- Auer rods in myeloblasts.
- Decreased neutrophils in bone marrow.
As is found in rare cases of AML with t(8;21), the bone marrow blast percentage in this AML is occasionally less than 20%.
Both inv(16)(p13;q22) and t(16;16)(p13;q22) result in the fusion of the CBFB gene at 16q22 to the smooth muscle MYH11 gene at 16p13, thereby forming the CBFB::MYH11 fusion gene .[11] The use of FISH and RT–PCR methods is sometimes necessary to document this fusion gene because its presence is not always documented by traditional cytogenetics banding techniques.[20] Similar to AML with t(8;21), patients with the CBFB::MYH11 fusion gene achieve higher CR rates and long-term survival when treated with high-dose cytarabine in the postremission setting.[12,13,15] Unlike AML with t(8;21), the prognostic relevance of KIT mutations is unclear.[21]
APL with PML::RARA
APL is defined by the presence of the PML::RARA fusion protein, typically a result of t(15;17)(q22;q12), but can be cryptic or result from complex cytogenetic rearrangements other than t(15;17)(q22;q12). It is also an AML in which promyelocytes are the dominant leukemic cell type. APL exists as two subtypes, hypergranular or typical APL and microgranular or hypogranular APL. APL comprises 5% to 8% of cases of AML and occurs predominately in adults in midlife.[5] Both typical and microgranular APL are commonly associated with disseminated intravascular coagulation (DIC).[22,23] In microgranular APL, unlike typical APL, the leukocyte count can be very high with a rapid doubling time.[5]
Common morphological features of typical APL include the following:
- Kidney-shaped or bilobed nuclei.
- Cytoplasm densely packed with large granules (bright pink, red, or purple in Romanowsky stains).
- Bundles of Auer rods within the cytoplasm (faggot cells).
- Larger Auer rods than in other types of AML.
- Strongly positive myeloperoxidase (MPO) reaction in all leukemic promyelocytes.
- Only occasional leukemic promyelocytes in the blood.
Common morphological features of microgranular APL include the following:
- Bilobed nuclear shape.
- Apparent scarce or absent granules (submicroscopic azurophilic granules).
- Small number of abnormal promyelocytes with visible granules and/or bundles of Auer rods (faggot cells).
- High leukocyte count in the peripheral blood.
- Strongly positive MPO reaction in all leukemic promyelocytes.
In APL, the RARA gene on 17q12 fuses with a nuclear regulatory factor on 15q22 (PML gene) resulting in a PML::RARA gene fusion transcript.[24,25,26] Rare cases of cryptic or masked t(15;17) lack typical cytogenetic findings and involve complex variant translocations or submicroscopic insertion of the RARA gene into the PML gene, leading to the expression of the PML::RARA fusion transcript.[5] FISH and/or RT–PCR methods may be required to unmask these cryptic genetic rearrangements.[27,28] In approximately 1% of the patients with APL, variant chromosomal aberrations may be found in which the RARA gene is fused with other genes.[29] Variant translocations involving the RARA gene include t(11;17)(q23;q21), t(5;17)(q32;q12), and t(11;17)(q13;q21).[5]
APL has a specific sensitivity to treatment with all-trans retinoic acid (ATRA, tretinoin), which acts as a differentiating agent.[30,31,32] High CR rates and long-term disease-free survival in APL may be obtained by combining ATRA treatment with chemotherapy,[33] or in a chemotherapy-free regimen with arsenic trioxide.[34]
AML with t(9;11)(p21.3;q23.3), MLLT3::KMT2A
AML with 11q23 abnormalities comprises 5% to 6% of cases of AML and is typically associated with monocytic features. This type of AML is more common in children. Two clinical subgroups who have a high frequency of AML with 11q23 abnormalities are infants with AML and patients with therapy-related AML, usually occurring after treatment with DNA topoisomerase inhibitors. Patients may present with DIC and extramedullary monocytic sarcomas and/or tissue infiltration (gingiva, skin).[5]
Common morphological features include the following:
- Monoblasts and promonocytes predominate in the bone marrow.
- Monoblasts and promonocytes with strong, positive nonspecific-esterase reactions.
The MLLT3 gene on 11q23, an epigenetic regulator, is involved in translocations with approximately 135 different rearrangements having been identified so far.[35] Genes other than MLLT3 may be involved in 11q23 abnormalities.[36] FISH may be required to detect genetic abnormalities involving MLL.[36,37,38] In general, risk categories and prognoses for individual 11q23 translocations are difficult to determine because of the lack of studies involving significant numbers of patients; however, patients with t(11;19)(q23;p13.1) have been reported to have poor outcomes.[13]
AML with t(6;9)(p23;q34.1),DEK::NUP214
The t(6;9) translocation leads to the formation of a leukemia-associated DEK::NUP214 fusion protein and accounts for approximately 1% of AML cases.[39,40,41] NUP214 is a component of the nuclear pore complex. This subgroup of AML has been associated with a poor prognosis.[39,42,43]
AML with inv(3)(q21.3;q26.2) or t(3;3)(q21.3;q26.2),GATA2,MECOM
The inv(3) abnormality or t(3;3) translocation occur infrequently and account for approximately 1% of all AML cases.[41]MECOM at chromosome 3q26 codes for two proteins, EVI1 and MDS1-EVI1, both of which are transcription regulators. The inv(3) and t(3;3) abnormalities do not lead to a fusion gene, rather they reposition the distal GATA2 enhancer, resulting in overexpression of EVI1, and simultaneously confer GATA2 haploinsufficiency.[44,45] These abnormalities are associated with poor prognosis.[15,46,47] Abnormalities involving MECOM can be detected in some AML cases with other 3q abnormalities and are also associated with poor prognosis.
AML (megakaryoblastic) with t(1;22)(p13.3;q13.3),RBM15::MKL1
The t(1;22)(p13;q13) translocation that produces the RBM15::MKL1 fusion gene is an uncommon driver of pediatric AML (<1% of pediatric AML) and is restricted to acute megakaryocytic leukemia. For more information, see Childhood Acute Myeloid Leukemia Treatment.
AML withBCR::ABL1(provisional entity)
This provisional entity was added by the WHO in 2016 in an effort to recognize that patients with the BCR::ABL1 fusion protein should be treated with a tyrosine kinase inhibitor.[10] However, this entity is very difficult to distinguish from chronic myelogenous leukemia (CML) in blast phase (BP-CML). Loss of IKZF1 and/or CDKN2A may help distinguish true cases of AML with BCR::ABL1 from BP-CML.[48] For more information, see Chronic Myeloid Leukemia Treatment.
AML with mutatedNPM1
NPM1 is a protein that has been linked to ribosomal protein assembly and transport and is also a molecular chaperone involved in preventing protein aggregation in the nucleolus. Immunohistochemical methods can be used to accurately identify patients with NPM1 mutations by the demonstration of cytoplasmic localization of NPM.[49] Mutations in the NPM1 protein diminish its nuclear localization and lead to impaired hematopoietic differentiation. They are primarily associated with a normal karyotype (50%), and less commonly seen in conjunction with an abnormal karyotype (<10%), or complex karyotype (<3%).[50,51,52] The presence of an NPM1 mutation confers improved prognosis in the absence of FLT3–internal tandem duplication (ITD) mutations.[50,53,54]
AML with biallelic mutations ofCEBPA
In adults younger than 60 years, 10% to 15% of cytogenetically normal AML cases have mutations in CEBPA.[53,55] The CEBPA gene is located on chromosome 19 and encodes a transcription factor that coordinates myeloid differentiation and cellular growth arrest.[56]
Outcomes for patients with AML with CEBPA mutations are relatively favorable and similar to that of patients with core-binding factor leukemias.[53,57] Studies have demonstrated that CEBPA double-mutant, but not single-mutant, AML is independently associated with a favorable prognosis,[55,58,59,60] leading to the WHO 2016 revision that requires biallelic mutations for the disease definition.[10]
AML with mutatedRUNX1(provisional entity)
AML with mutated RUNX1, which is a provisional entity in the 2016 WHO classification of AML and related neoplasms, denotes a distinct population of de novo AML without myelodysplastic syndrome (MDS)-related features.[61] Mutations in RUNX1 are associated with a high risk of treatment failure.[62,63,64]
AML With Myelodysplasia-Related Features
AML with myelodysplasia-related features is characterized by 20% or more blasts in the blood or bone marrow and dysplasia in two or more myeloid cell lines, generally including megakaryocytes.[5] To make the diagnosis, dysplasia must be present in 50% or more of the cells of at least two lineages and must be present in a pretreatment bone marrow specimen or must have the presence of an MDS-related cytogenetic abnormality.[5] AML with myelodysplasia-related features may occur de novo or after MDS or a myelodysplastic/myeloproliferative neoplasm overlap. The diagnostic terminology AML with myelodysplasia-related features evolving from a myelodysplastic syndrome should be used when an MDS precedes AML.[5] In the presence of a mutation in NPM1 or biallelic mutations of CEBPA, the presence of multilineage dysplasia alone will not classify a case as AML with myelodysplasia-related changes.[5] For more information, see Myelodysplastic Syndromes Treatment and Myelodysplastic/Myeloproliferative Neoplasms Treatment.
AML with myelodysplasia-related features occurs primarily in older patients.[5] Patients with AML with myelodysplasia-related features frequently present with severe pancytopenia.
Common morphological features include the following:
- Multilineage dysplasia in the blood or bone marrow.
- Dysplasia in 50% or more of the cells of two or more cell lines.
- Dysgranulopoiesis (neutrophils with hypogranular cytoplasm, hyposegmented nuclei or bizarrely segmented nuclei).
- Dyserythropoiesis (megaloblastic nuclei, karyorrhexis, or multinucleation of erythroid precursors and ringed sideroblasts).
- Dysmegakaryopoiesis (micromegakaryocytes and normal size or large megakaryocytes with monolobed or multiple separated nuclei).
Chromosome abnormalities observed in AML with myelodysplasia-related features are similar to those found in MDS and frequently involve gain or loss of major segments of certain chromosomes, predominately chromosomes 5 and/or 7. The probability of achieving a CR has been reported to be affected adversely by a diagnosis of AML with myelodysplasia-related features.[65,66,67]
Therapy-Related Myeloid Neoplasms
Therapy-related myeloid neoplasms (t-MN) include AML (t-AML) and MDS (t-MDS) that arise secondary to cytotoxic chemotherapy and/or radiation therapy.[5] The therapy-related (or secondary) MDS are included because of their close clinicopathological relationships to therapy-related AML. Although these therapy-related disorders can be distinguished by the specific mutagenic agents involved, this distinction may be difficult to make because of the frequent overlapping use of multiple potentially mutagenic agents in treating cancer.[68] Because the associated cytogenetic abnormality, not the mutagenetic agent, determines prognosis and treatment it should be noted in the diagnosis.[10]
Given that t-MN has been associated with germline mutations in cancer susceptibility genes, consideration for germline testing or genetic counseling is warranted in those with strong family histories.[69]
Alkylating agent-related t-MN
The alkylating agent/radiation-related acute leukemias and myelodysplastic syndromes typically occur 5 to 6 years after exposure to the mutagenic agent, with a reported range of approximately 10 to 192 months.[70,71] The risk of occurrence is related to both the total cumulative dose of the alkylating agent and the age of the patient.
Cytogenetic abnormalities have been observed in more than 90% of cases of t-MN and commonly include chromosomes 5 and/or 7.[70,72,73] Complex chromosomal abnormalities (≥3 distinct abnormalities) are the most common finding.[68,72,73,74]
Topoisomerase II inhibitor-related t-MN
Topoisomerase II inhibitor-related t-MN occurs in patients treated with topoisomerase II inhibitors. The agents implicated are the epipodophyllotoxins etoposide and teniposide and the anthracyclines doxorubicin and 4-epi-doxorubicin.[70] The mean latency period from the time of institution of the causative therapy to the development of t-MN is approximately 2 years.[75]
As with alkylating agent/radiation-related t-MN, the cytogenetic abnormalities are often complex.[68,72,73,74] The predominant cytogenetic finding involves chromosome 11q23 and the MLL gene.[68,76]
AML, Not Otherwise Specified (NOS)
Cases of AML that do not fulfill the criteria for AML with recurrent genetic abnormalities, AML with myelodysplasia-related features, or t-MN fall within the category of AML, NOS.[10] As mentioned before, the subcategories of AML, NOS lack prognostic significance when the mutation status of NPM1 and CEBPA are known.[9] Classification in this subset of AML is based on leukemic cell features of morphology, cytochemistry, and maturation (i.e., the FAB classification system) and include the following:[5]
- AML with minimal differentiation.
- AML without maturation.
- AML with maturation.
- Acute myelomonocytic leukemia.
- Acute monoblastic/monocytic leukemia.
- Pure erythroid leukemia.
- Acute megakaryoblastic leukemia.
- Acute basophilic leukemia.
- Acute panmyelosis with myelofibrosis.
Myeloid Sarcoma
Myeloid sarcoma (also known as extramedullary myeloid tumor, granulocytic sarcoma, and chloroma) is a tumor mass that consists of myeloblasts or immature myeloid cells, occurring in an extramedullary site.[5] Development of myeloid sarcoma has been reported in 2% to 8% of patients with AML.[77] Clinical features include occurrence common in subperiosteal bone structures of the skull, paranasal sinuses, sternum, ribs, vertebrae, and pelvis; lymph nodes, skin, mediastinum, small intestine, and the epidural space; and occurrence de novo or concomitant with AML or a myeloproliferative disorder.[10,77,78]
Morphological and cytochemical features include the following:
- Granulocytic sarcoma composed of myeloblasts, neutrophils, and neutrophil precursors with three subtypes based on degree of maturation (i.e., blastic, immature, and differentiated).
- Monoblastic sarcoma preceding or occurring simultaneously with acute monoblastic leukemia.
- Tumors with trilineage hematopoiesis occurring with transformation of chronic myeloproliferative disorders.
- Myeloblasts and neutrophils that are positive for MPO.
- Neutrophils that are positive for naphthol ASD chloroacetate esterase.
Immunophenotyping with antibodies to MPO, lysozyme, and chloroacetate is critical to the diagnosis of these lesions.[5] The myeloblasts in granulocytic sarcomas express myeloid-associated antigens (CD13, CD33, CD117, and MPO). The monoblasts in monoblastic sarcomas express acute monoblastic leukemia antigens (CD14, CD116, and CD11c) and usually react with antibodies to lysozyme and CD68. The main differential diagnosis includes non-Hodgkin lymphoma of the lymphoblastic type, Burkitt lymphoma, large-cell lymphoma, and small, round-cell tumors, especially in children (e.g., neuroblastoma, rhabdomyosarcoma, Ewing/primitive neuroectodermal tumors, and medulloblastoma). When able, FISH for common chromosomal abnormalities should be completed, as well as molecular studies to refine diagnosis and aid in prognosis.
No unique chromosomal abnormalities are associated with myeloid sarcoma.[77,79] The presence of myeloid sarcoma in patients with the otherwise good-risk t(8;21) AML may be associated with a lower CR rate and decreased remission duration.[80] Myeloid sarcoma occurring in the setting of MDS or myeloproliferative disorder is equivalent to blast transformation (progression to AML). In the case of AML, the prognosis is that of the underlying leukemia.[10] Although the initial presentation of myeloid sarcoma may appear to be isolated, it is a partial manifestation of a systemic disease and should be treated with intensive chemotherapy.[77,78,81,82]
Myeloid Proliferations Related to Down Syndrome
For more information about TAM and myeloid leukemia associated with Down syndrome, see Childhood Myeloid Proliferations Associated With Down Syndrome Treatment.
Acute Leukemias of Ambiguous Lineage
Acute leukemias of ambiguous lineage are rare types of acute leukemia in which the morphological, cytochemical, and immunophenotypic features of the blast population do not allow classification in myeloid or lymphoid categories; or the types have morphological and/or immunophenotypic features of both myeloid and lymphoid cells or both B and T lineages (i.e., acute bilineal leukemia and acute biphenotypic leukemia).[10,83,84]
They include the following subcategories:[5]
- Acute undifferentiated leukemia.
- Mixed phenotype acute leukemia (MPAL) with t(9;22)(q34.1;q11.2); BCR::ABL1.
- MPAL with t(v;11q23.3); KMT2A rearranged.
- MPAL, B/myeloid, NOS.
- MPAL, T/myeloid, NOS.
The diagnosis of MPAL is made in leukemias with expression of antigens of more than one lineage:[5]
Table 1. Mixed Phenotype Acute Leukemia Diagnostic Criteria
| Diagnosis |
Criteria |
| MPO = myeloperoxidase. |
| Myeloid Lineage |
MPO (flow cytometry, immunohistochemistry, or cytochemistry) or monocytic differentiation (≥ 2 of the following: nonspecific esterase cytochemistry, CD11c, CD14, CD64, lysozyme). |
| T-cell Lineage |
Strong cytoplasmic CD3 (with antibodies to CD3 epsilon chain) or surface CD3. |
| B-cell Lineage |
Strong CD19 with ≥1 of the following strongly expressed: cytoplasmic CD79a, cCD22, or CD10; or weak CD19 with at least two of the following strongly expressed: CD79a, cCD22, or CD10. |
Cytogenetic abnormalities are observed in a high percentage of acute leukemias of ambiguous lineage.[85,86,87,88] Approximately 33% of cases have the Philadelphia chromosome, and some cases are associated with t(4;11)(q21;q23) or other 11q23 abnormalities. In general, the prognosis appears to be unfavorable. The occurrence of 11q23 abnormalities or BCR::ABL1 are especially unfavorable prognostic indicators;[86,89,90] however, preliminary results indicate that tyrosine kinase inhibitors can be used successfully.[91,92]
References:
-
Brunning RD, Matutes E, Harris NL, et al.: Acute myeloid leukaemia: introduction. In: Jaffe ES, Harris NL, Stein H, et al., eds.: Pathology and Genetics of Tumours of Haematopoietic and Lymphoid Tissues. IARC Press, 2001. World Health Organization Classification of Tumours, 3, pp 77-80.
-
Bennett JM, Catovsky D, Daniel MT, et al.: Proposed revised criteria for the classification of acute myeloid leukemia. A report of the French-American-British Cooperative Group. Ann Intern Med 103 (4): 620-5, 1985.
-
Cheson BD, Cassileth PA, Head DR, et al.: Report of the National Cancer Institute-sponsored workshop on definitions of diagnosis and response in acute myeloid leukemia. J Clin Oncol 8 (5): 813-9, 1990.
-
Bennett JM, Catovsky D, Daniel MT, et al.: Proposals for the classification of the acute leukaemias. French-American-British (FAB) co-operative group. Br J Haematol 33 (4): 451-8, 1976.
-
Swerdlow SH, Campo E, Harris NL, et al., eds.: WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. 4th rev. ed. International Agency for Research on Cancer, 2017.
-
Jaffe ES, Harris NL, Stein H, et al., eds.: Pathology and Genetics of Tumours of Haematopoietic and Lymphoid Tissues. IARC Press, 2001. World Health Organization Classification of Tumours, 3.
-
Hasle H, Niemeyer CM, Chessells JM, et al.: A pediatric approach to the WHO classification of myelodysplastic and myeloproliferative diseases. Leukemia 17 (2): 277-82, 2003.
-
Arber DA, Vardiman JW, Brunning RD: Acute myeloid leukaemia with recurrent genetic abnormalities. In: Swerdlow SH, Campo E, Harris NL, et al., eds.: WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. 4th ed. International Agency for Research on Cancer, 2008, pp 110-23.
-
Walter RB, Othus M, Burnett AK, et al.: Significance of FAB subclassification of "acute myeloid leukemia, NOS" in the 2008 WHO classification: analysis of 5848 newly diagnosed patients. Blood 121 (13): 2424-31, 2013.
-
Arber DA, Orazi A, Hasserjian R, et al.: The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood 127 (20): 2391-405, 2016.
-
Caligiuri MA, Strout MP, Gilliland DG: Molecular biology of acute myeloid leukemia. Semin Oncol 24 (1): 32-44, 1997.
-
Bloomfield CD, Lawrence D, Byrd JC, et al.: Frequency of prolonged remission duration after high-dose cytarabine intensification in acute myeloid leukemia varies by cytogenetic subtype. Cancer Res 58 (18): 4173-9, 1998.
-
Byrd JC, Mrózek K, Dodge RK, et al.: Pretreatment cytogenetic abnormalities are predictive of induction success, cumulative incidence of relapse, and overall survival in adult patients with de novo acute myeloid leukemia: results from Cancer and Leukemia Group B (CALGB 8461). Blood 100 (13): 4325-36, 2002.
-
Palmieri S, Sebastio L, Mele G, et al.: High-dose cytarabine as consolidation treatment for patients with acute myeloid leukemia with t(8;21). Leuk Res 26 (6): 539-43, 2002.
-
Grimwade D, Walker H, Oliver F, et al.: The importance of diagnostic cytogenetics on outcome in AML: analysis of 1,612 patients entered into the MRC AML 10 trial. The Medical Research Council Adult and Children's Leukaemia Working Parties. Blood 92 (7): 2322-33, 1998.
-
Downing JR: The AML1-ETO chimaeric transcription factor in acute myeloid leukaemia: biology and clinical significance. Br J Haematol 106 (2): 296-308, 1999.
-
Schlenk RF, Benner A, Krauter J, et al.: Individual patient data-based meta-analysis of patients aged 16 to 60 years with core binding factor acute myeloid leukemia: a survey of the German Acute Myeloid Leukemia Intergroup. J Clin Oncol 22 (18): 3741-50, 2004.
-
Duployez N, Marceau-Renaut A, Boissel N, et al.: Comprehensive mutational profiling of core binding factor acute myeloid leukemia. Blood 127 (20): 2451-9, 2016.
-
Marlton P, Keating M, Kantarjian H, et al.: Cytogenetic and clinical correlates in AML patients with abnormalities of chromosome 16. Leukemia 9 (6): 965-71, 1995.
-
Poirel H, Radford-Weiss I, Rack K, et al.: Detection of the chromosome 16 CBF beta-MYH11 fusion transcript in myelomonocytic leukemias. Blood 85 (5): 1313-22, 1995.
-
Döhner K, Paschka P: Intermediate-risk acute myeloid leukemia therapy: current and future. Hematology Am Soc Hematol Educ Program 2014 (1): 34-43, 2014.
-
Kwaan HC, Wang J, Boggio LN: Abnormalities in hemostasis in acute promyelocytic leukemia. Hematol Oncol 20 (1): 33-41, 2002.
-
Barbui T, Falanga A: Disseminated intravascular coagulation in acute leukemia. Semin Thromb Hemost 27 (6): 593-604, 2001.
-
de Thé H, Chomienne C, Lanotte M, et al.: The t(15;17) translocation of acute promyelocytic leukaemia fuses the retinoic acid receptor alpha gene to a novel transcribed locus. Nature 347 (6293): 558-61, 1990.
-
Melnick A, Licht JD: Deconstructing a disease: RARalpha, its fusion partners, and their roles in the pathogenesis of acute promyelocytic leukemia. Blood 93 (10): 3167-215, 1999.
-
Kayser S, Schlenk RF, Platzbecker U: Management of patients with acute promyelocytic leukemia. Leukemia 32 (6): 1277-1294, 2018.
-
Lo Coco F, Diverio D, Falini B, et al.: Genetic diagnosis and molecular monitoring in the management of acute promyelocytic leukemia. Blood 94 (1): 12-22, 1999.
-
Zaccaria A, Valenti A, Toschi M, et al.: Cryptic translocation of PML/RARA on 17q. A rare event in acute promyelocytic leukemia. Cancer Genet Cytogenet 138 (2): 169-73, 2002.
-
Jansen JH, Löwenberg B: Acute promyelocytic leukemia with a PLZF-RARalpha fusion protein. Semin Hematol 38 (1): 37-41, 2001.
-
Castaigne S, Chomienne C, Daniel MT, et al.: All-trans retinoic acid as a differentiation therapy for acute promyelocytic leukemia. I. Clinical results. Blood 76 (9): 1704-9, 1990.
-
Tallman MS, Andersen JW, Schiffer CA, et al.: All-trans-retinoic acid in acute promyelocytic leukemia. N Engl J Med 337 (15): 1021-8, 1997.
-
Tallman MS, Andersen JW, Schiffer CA, et al.: All-trans retinoic acid in acute promyelocytic leukemia: long-term outcome and prognostic factor analysis from the North American Intergroup protocol. Blood 100 (13): 4298-302, 2002.
-
Fenaux P, Chastang C, Chevret S, et al.: A randomized comparison of all transretinoic acid (ATRA) followed by chemotherapy and ATRA plus chemotherapy and the role of maintenance therapy in newly diagnosed acute promyelocytic leukemia. The European APL Group. Blood 94 (4): 1192-200, 1999.
-
Lo-Coco F, Avvisati G, Vignetti M, et al.: Retinoic acid and arsenic trioxide for acute promyelocytic leukemia. N Engl J Med 369 (2): 111-21, 2013.
-
Meyer C, Burmeister T, Gröger D, et al.: The MLL recombinome of acute leukemias in 2017. Leukemia 32 (2): 273-284, 2018.
-
Giugliano E, Rege-Cambrin G, Scaravaglio P, et al.: Two new translocations involving the 11q23 region map outside the MLL locus in myeloid leukemias. Haematologica 87 (10): 1014-20, 2002.
-
König M, Reichel M, Marschalek R, et al.: A highly specific and sensitive fluorescence in situ hybridization assay for the detection of t(4;11)(q21;q23) and concurrent submicroscopic deletions in acute leukaemias. Br J Haematol 116 (4): 758-64, 2002.
-
Kim HJ, Cho HI, Kim EC, et al.: A study on 289 consecutive Korean patients with acute leukaemias revealed fluorescence in situ hybridization detects the MLL translocation without cytogenetic evidence both initially and during follow-up. Br J Haematol 119 (4): 930-9, 2002.
-
Ageberg M, Drott K, Olofsson T, et al.: Identification of a novel and myeloid specific role of the leukemia-associated fusion protein DEK-NUP214 leading to increased protein synthesis. Genes Chromosomes Cancer 47 (4): 276-87, 2008.
-
Shiba N, Ichikawa H, Taki T, et al.: NUP98-NSD1 gene fusion and its related gene expression signature are strongly associated with a poor prognosis in pediatric acute myeloid leukemia. Genes Chromosomes Cancer 52 (7): 683-93, 2013.
-
Döhner H, Estey E, Grimwade D, et al.: Diagnosis and management of AML in adults: 2017 ELN recommendations from an international expert panel. Blood 129 (4): 424-447, 2017.
-
Slovak ML, Gundacker H, Bloomfield CD, et al.: A retrospective study of 69 patients with t(6;9)(p23;q34) AML emphasizes the need for a prospective, multicenter initiative for rare 'poor prognosis' myeloid malignancies. Leukemia 20 (7): 1295-7, 2006.
-
Alsabeh R, Brynes RK, Slovak ML, et al.: Acute myeloid leukemia with t(6;9) (p23;q34): association with myelodysplasia, basophilia, and initial CD34 negative immunophenotype. Am J Clin Pathol 107 (4): 430-7, 1997.
-
Gröschel S, Sanders MA, Hoogenboezem R, et al.: A single oncogenic enhancer rearrangement causes concomitant EVI1 and GATA2 deregulation in leukemia. Cell 157 (2): 369-81, 2014.
-
Yamazaki H, Suzuki M, Otsuki A, et al.: A remote GATA2 hematopoietic enhancer drives leukemogenesis in inv(3)(q21;q26) by activating EVI1 expression. Cancer Cell 25 (4): 415-27, 2014.
-
Mrózek K, Heerema NA, Bloomfield CD: Cytogenetics in acute leukemia. Blood Rev 18 (2): 115-36, 2004.
-
Lugthart S, Gröschel S, Beverloo HB, et al.: Clinical, molecular, and prognostic significance of WHO type inv(3)(q21q26.2)/t(3;3)(q21;q26.2) and various other 3q abnormalities in acute myeloid leukemia. J Clin Oncol 28 (24): 3890-8, 2010.
-
Nacheva EP, Grace CD, Brazma D, et al.: Does BCR/ABL1 positive acute myeloid leukaemia exist? Br J Haematol 161 (4): 541-50, 2013.
-
Falini B, Martelli MP, Bolli N, et al.: Immunohistochemistry predicts nucleophosmin (NPM) mutations in acute myeloid leukemia. Blood 108 (6): 1999-2005, 2006.
-
Falini B, Mecucci C, Tiacci E, et al.: Cytoplasmic nucleophosmin in acute myelogenous leukemia with a normal karyotype. N Engl J Med 352 (3): 254-66, 2005.
-
Falini B, Nicoletti I, Martelli MF, et al.: Acute myeloid leukemia carrying cytoplasmic/mutated nucleophosmin (NPMc+ AML): biologic and clinical features. Blood 109 (3): 874-85, 2007.
-
Falini B, Martelli MP, Bolli N, et al.: Acute myeloid leukemia with mutated nucleophosmin (NPM1): is it a distinct entity? Blood 117 (4): 1109-20, 2011.
-
Schlenk RF, Döhner K, Krauter J, et al.: Mutations and treatment outcome in cytogenetically normal acute myeloid leukemia. N Engl J Med 358 (18): 1909-18, 2008.
-
Gale RE, Green C, Allen C, et al.: The impact of FLT3 internal tandem duplication mutant level, number, size, and interaction with NPM1 mutations in a large cohort of young adult patients with acute myeloid leukemia. Blood 111 (5): 2776-84, 2008.
-
Taskesen E, Bullinger L, Corbacioglu A, et al.: Prognostic impact, concurrent genetic mutations, and gene expression features of AML with CEBPA mutations in a cohort of 1182 cytogenetically normal AML patients: further evidence for CEBPA double mutant AML as a distinctive disease entity. Blood 117 (8): 2469-75, 2011.
-
Nerlov C: C/EBPalpha mutations in acute myeloid leukaemias. Nat Rev Cancer 4 (5): 394-400, 2004.
-
Marcucci G, Maharry K, Radmacher MD, et al.: Prognostic significance of, and gene and microRNA expression signatures associated with, CEBPA mutations in cytogenetically normal acute myeloid leukemia with high-risk molecular features: a Cancer and Leukemia Group B Study. J Clin Oncol 26 (31): 5078-87, 2008.
-
Wouters BJ, Löwenberg B, Erpelinck-Verschueren CA, et al.: Double CEBPA mutations, but not single CEBPA mutations, define a subgroup of acute myeloid leukemia with a distinctive gene expression profile that is uniquely associated with a favorable outcome. Blood 113 (13): 3088-91, 2009.
-
Dufour A, Schneider F, Metzeler KH, et al.: Acute myeloid leukemia with biallelic CEBPA gene mutations and normal karyotype represents a distinct genetic entity associated with a favorable clinical outcome. J Clin Oncol 28 (4): 570-7, 2010.
-
Fasan A, Haferlach C, Alpermann T, et al.: The role of different genetic subtypes of CEBPA mutated AML. Leukemia 28 (4): 794-803, 2014.
-
Schnittger S, Dicker F, Kern W, et al.: RUNX1 mutations are frequent in de novo AML with noncomplex karyotype and confer an unfavorable prognosis. Blood 117 (8): 2348-57, 2011.
-
Tang JL, Hou HA, Chen CY, et al.: AML1/RUNX1 mutations in 470 adult patients with de novo acute myeloid leukemia: prognostic implication and interaction with other gene alterations. Blood 114 (26): 5352-61, 2009.
-
Mendler JH, Maharry K, Radmacher MD, et al.: RUNX1 mutations are associated with poor outcome in younger and older patients with cytogenetically normal acute myeloid leukemia and with distinct gene and MicroRNA expression signatures. J Clin Oncol 30 (25): 3109-18, 2012.
-
Gaidzik VI, Bullinger L, Schlenk RF, et al.: RUNX1 mutations in acute myeloid leukemia: results from a comprehensive genetic and clinical analysis from the AML study group. J Clin Oncol 29 (10): 1364-72, 2011.
-
Díaz-Beyá M, Rozman M, Pratcorona M, et al.: The prognostic value of multilineage dysplasia in de novo acute myeloid leukemia patients with intermediate-risk cytogenetics is dependent on NPM1 mutational status. Blood 116 (26): 6147-8, 2010.
-
Rozman M, Navarro JT, Arenillas L, et al.: Multilineage dysplasia is associated with a poorer prognosis in patients with de novo acute myeloid leukemia with intermediate-risk cytogenetics and wild-type NPM1. Ann Hematol 93 (10): 1695-703, 2014.
-
Weinberg OK, Seetharam M, Ren L, et al.: Clinical characterization of acute myeloid leukemia with myelodysplasia-related changes as defined by the 2008 WHO classification system. Blood 113 (9): 1906-8, 2009.
-
Smith SM, Le Beau MM, Huo D, et al.: Clinical-cytogenetic associations in 306 patients with therapy-related myelodysplasia and myeloid leukemia: the University of Chicago series. Blood 102 (1): 43-52, 2003.
-
Churpek JE, Marquez R, Neistadt B, et al.: Inherited mutations in cancer susceptibility genes are common among survivors of breast cancer who develop therapy-related leukemia. Cancer 122 (2): 304-11, 2016.
-
Brunning RD, Matutes E, Flandrin G, et al.: Acute myeloid leukaemias and myelodysplastic syndromes, therapy related. In: Jaffe ES, Harris NL, Stein H, et al., eds.: Pathology and Genetics of Tumours of Haematopoietic and Lymphoid Tissues. IARC Press, 2001. World Health Organization Classification of Tumours, 3, pp 89-91.
-
Ellis M, Ravid M, Lishner M: A comparative analysis of alkylating agent and epipodophyllotoxin-related leukemias. Leuk Lymphoma 11 (1-2): 9-13, 1993.
-
Olney HJ, Mitelman F, Johansson B, et al.: Unique balanced chromosome abnormalities in treatment-related myelodysplastic syndromes and acute myeloid leukemia: report from an international workshop. Genes Chromosomes Cancer 33 (4): 413-23, 2002.
-
Mauritzson N, Albin M, Rylander L, et al.: Pooled analysis of clinical and cytogenetic features in treatment-related and de novo adult acute myeloid leukemia and myelodysplastic syndromes based on a consecutive series of 761 patients analyzed 1976-1993 and on 5098 unselected cases reported in the literature 1974-2001. Leukemia 16 (12): 2366-78, 2002.
-
Pedersen-Bjergaard J, Andersen MK, Christiansen DH, et al.: Genetic pathways in therapy-related myelodysplasia and acute myeloid leukemia. Blood 99 (6): 1909-12, 2002.
-
Leone G, Voso MT, Sica S, et al.: Therapy related leukemias: susceptibility, prevention and treatment. Leuk Lymphoma 41 (3-4): 255-76, 2001.
-
Bloomfield CD, Archer KJ, Mrózek K, et al.: 11q23 balanced chromosome aberrations in treatment-related myelodysplastic syndromes and acute leukemia: report from an international workshop. Genes Chromosomes Cancer 33 (4): 362-78, 2002.
-
Yamauchi K, Yasuda M: Comparison in treatments of nonleukemic granulocytic sarcoma: report of two cases and a review of 72 cases in the literature. Cancer 94 (6): 1739-46, 2002.
-
Yilmaz AF, Saydam G, Sahin F, et al.: Granulocytic sarcoma: a systematic review. Am J Blood Res 3 (4): 265-70, 2013.
-
Brunning RD, Matutes E, Flandrin G, et al.: Acute myeloid leukaemia not otherwise categorised. In: Jaffe ES, Harris NL, Stein H, et al., eds.: Pathology and Genetics of Tumours of Haematopoietic and Lymphoid Tissues. IARC Press, 2001. World Health Organization Classification of Tumours, 3, pp 91-105.
-
Byrd JC, Weiss RB, Arthur DC, et al.: Extramedullary leukemia adversely affects hematologic complete remission rate and overall survival in patients with t(8;21)(q22;q22): results from Cancer and Leukemia Group B 8461. J Clin Oncol 15 (2): 466-75, 1997.
-
Hayashi T, Kimura M, Satoh S, et al.: Early detection of AML1/MTG8 fusion mRNA by RT-PCR in the bone marrow cells from a patient with isolated granulocytic sarcoma. Leukemia 12 (9): 1501-3, 1998.
-
Imrie KR, Kovacs MJ, Selby D, et al.: Isolated chloroma: the effect of early antileukemic therapy. Ann Intern Med 123 (5): 351-3, 1995.
-
Matutes E, Pickl WF, Van't Veer M, et al.: Mixed-phenotype acute leukemia: clinical and laboratory features and outcome in 100 patients defined according to the WHO 2008 classification. Blood 117 (11): 3163-71, 2011.
-
van den Ancker W, Terwijn M, Westers TM, et al.: Acute leukemias of ambiguous lineage: diagnostic consequences of the WHO2008 classification. Leukemia 24 (7): 1392-6, 2010.
-
Hanson CA, Abaza M, Sheldon S, et al.: Acute biphenotypic leukaemia: immunophenotypic and cytogenetic analysis. Br J Haematol 84 (1): 49-60, 1993.
-
Legrand O, Perrot JY, Simonin G, et al.: Adult biphenotypic acute leukaemia: an entity with poor prognosis which is related to unfavourable cytogenetics and P-glycoprotein over-expression. Br J Haematol 100 (1): 147-55, 1998.
-
Carbonell F, Swansbury J, Min T, et al.: Cytogenetic findings in acute biphenotypic leukaemia. Leukemia 10 (8): 1283-7, 1996.
-
Pane F, Frigeri F, Camera A, et al.: Complete phenotypic and genotypic lineage switch in a Philadelphia chromosome-positive acute lymphoblastic leukemia. Leukemia 10 (4): 741-5, 1996.
-
Brunning RD, Matutes E, Borowitz M: Acute leukaemias of ambiguous lineage. In: Jaffe ES, Harris NL, Stein H, et al., eds.: Pathology and Genetics of Tumours of Haematopoietic and Lymphoid Tissues. IARC Press, 2001. World Health Organization Classification of Tumours, 3, pp 106-7.
-
Killick S, Matutes E, Powles RL, et al.: Outcome of biphenotypic acute leukemia. Haematologica 84 (8): 699-706, 1999.
-
Kawajiri C, Tanaka H, Hashimoto S, et al.: Successful treatment of Philadelphia chromosome-positive mixed phenotype acute leukemia by appropriate alternation of second-generation tyrosine kinase inhibitors according to BCR-ABL1 mutation status. Int J Hematol 99 (4): 513-8, 2014.
-
Shimizu H, Yokohama A, Hatsumi N, et al.: Philadelphia chromosome-positive mixed phenotype acute leukemia in the imatinib era. Eur J Haematol 93 (4): 297-301, 2014.
Treatment Option Overview for AML
Phases of Therapy
The treatment of patients with acute myeloid leukemia (AML) is based on whether the disease is newly diagnosed (previously untreated), in remission, or recurrent. Also, the intensity of the treatment and the patient's overall health status are considered when choosing a treatment approach. Successful treatment of AML requires the control of bone marrow and systemic disease, and specific treatment of central nervous system (CNS) disease, if present. The cornerstone of this strategy includes systemically administered combination chemotherapy. Because only 5% or fewer of patients with AML develop CNS disease, prophylactic treatment is not indicated.[1,2]
- Newly diagnosed (untreated): Untreated AML is defined as newly diagnosed leukemia that has not been previously treated. The initial treatment for patients with newly diagnosed AML is often induction therapy that aims to induce a remission. In patients with AML, a complete remission (CR) is defined as a normal peripheral blood cell count (absolute neutrophil count >1,000/mm3 and platelet count >100,000/mm3) and normocellular marrow with less than 5% blasts in the marrow and no signs or symptoms of the disease. In addition, no signs or symptoms are evident of CNS leukemia or other extramedullary infiltration.[3]
Modifications to the definition of CR have been proposed because some responses are deeper than a CR, and others may not meet all the criteria for a complete response. In addition, most AML patients meeting the criteria for CR have residual leukemia.[3]
Table 2. Treatment Response Categories for Newly Diagnosed Acute Myeloid Leukemia
| Response Category |
Definition |
| ANC = absolute neutrophil count; CR = complete remission; MLFS = morphological leukemia-free state; PR = partial remission; RT–qPCR = reverse transcription–quantitative polymerase chain reaction. |
| CR without measurable residual disease (CRMRD−) |
If studied pretreatment, CR with negativity for a genetic marker by RT–qPCR, or CR with negativity by multicolor flow cytometry. |
| CR |
Bone marrow blasts <5%; absence of circulating blasts and blasts with Auer rods; absence of extramedullary disease; ANC ≥1.0 × 109 /L (1,000/microL); platelet count ≥100 × 109 /L (100,000/microL). |
| CR with incomplete hematologic recovery (CRi) |
All CR criteria except for residual neutropenia (<1.0 × 109 /L [1,000/microL]) or thrombocytopenia (<100 × 109 /L [100,000/microL]). |
| MLFS |
Bone marrow blasts <5%; absence of blasts with Auer rods; absence of extramedullary disease; no hematologic recovery required. |
| PR |
All hematologic criteria of CR; decrease of bone marrow blast percentage to 5 to 25%; and decrease of pretreatment bone marrow blast percentage by at least 50%. |
- In remission: When patients are in a remission after induction chemotherapy, consolidation chemotherapy is given, with the aim of deepening the response and consolidating the remission. This phase of treatment is also called postremission (to maintain remission). Maintenance therapy is not included in most current treatment protocols and clinical trials. Postremission therapy appears to be effective when administered immediately after remission is achieved.[4]
- Persistent/recurrent disease: Despite intensive chemotherapy, some patients with newly diagnosed AML will not go into remission and have primary refractory disease. Also, some patients who are in a remission after induction and consolidation chemotherapy may have a return of their disease.[3] The rates of primary refractory disease and relapse vary with the age of the patient, genomic variants seen in the leukemia cells, and initial treatment given.
Table 3. Treatment Response Categories for Persistent/Recurrent Acute Myeloid Leukemia
| Response Category |
Definition |
| CR = complete remission; CRi = complete remission with incomplete hematologic recovery; MRD- = absence of measurable residual disease; MLFS = morphological leukemia-free state; PR = partial response; RT–qPCR = reverse transcription–quantitative polymerase chain reaction. |
| Primary refractory disease |
No CR or CRi after two courses of intensive induction treatment; excluding patients with death in aplasia or death due to an indeterminate cause. |
| Hematologic relapse (after CRMRD-, CR, CRi) |
Bone marrow blasts ≥5%; or reappearance of blasts in the blood; or development of extramedullary disease. |
| Molecular relapse (after CRMRD-) |
If studied pretreatment, reoccurrence of MRD as assessed by RT–qPCR or by multicolor flow cytometry. |
| Stable disease |
Absence of CRMRD-, CR, CRi, PR, MLFS; and criteria for progressive disease not met. |
| Progressive disease |
Evidence for an increase in bone marrow blast percentage and/or increase of absolute blast counts in the blood: |
| |
>50% increase in marrow blasts; or |
| |
>50% increase in peripheral blasts in the absence of differentiation syndrome; or |
| |
New extramedullary disease. |
Supportive Care During Therapy
Because myelosuppression is an anticipated consequence of both the leukemia and its treatment with chemotherapy, patients must be closely monitored during therapy. Facilities must be available for hematologic support with multiple blood fractions, including platelet transfusions, and for the treatment of related infectious complications.[5]
Transfusion therapy
Supportive care during remission induction treatment should routinely include red blood cell and platelet transfusions, when appropriate.[6,7] Rapid marrow ablation with consequent earlier marrow regeneration decreases morbidity and mortality. Randomized trials have shown similar outcomes for patients who received prophylactic platelet transfusions at a level of 10,000/mm3 rather than 20,000/mm3.[8] The incidence of platelet alloimmunization was similar among groups randomly assigned to receive pooled platelet concentrates from random donors; filtered, pooled platelet concentrates from random donors; ultraviolet B-irradiated, pooled platelet concentrates from random donors; or filtered platelets obtained by apheresis from single random donors.[9]
No good evidence exists to support granulocyte transfusions in the treatment of AML. A multicenter randomized trial (RING [NCT00627393]) was conducted to address the utility of granulocyte transfusions in the setting of infections.[10] There was no difference between the granulocyte and control arms for the composite primary end point of survival plus microbial response at 42 days after randomization. However, the power to detect a true beneficial effect was low because enrollment was half that of the planned study size.
Growth factors
The following growth factors have been studied in the treatment of AML:
- Colony-stimulating factors: Granulocyte colony–stimulating factor and granulocyte-macrophage colony–stimulating factor have been studied in an effort to shorten the period of granulocytopenia associated with leukemia treatment.[11] If used, these agents are administered after administration of chemotherapy. Although the use of growth factors decreases the time to neutrophil recovery by 2 to 5 days, and decreases rates of febrile neutropenia and duration of hospitalization, randomized clinical trials have not shown an impact of growth factors on overall survival and their cost-effectiveness has been inconsistently reported.[12,13] Use of growth factors is not routinely recommended in the remission induction setting.
- Erythropoiesis-stimulating agents: Anemia associated with the diagnosis of AML and induction chemotherapy is managed primarily with red blood transfusions. Unlike myelodysplastic syndromes, there is no role for the use of erythropoiesis stimulating agents (e.g., epoetin alfa and darbepoetin) during the treatment of AML.
- Thrombopoietin mimetics: Clinical trials have assessed the use of thrombopoietin mimetics in the treatment of AML. Eltrombopag as a single agent was tested in a multicenter, randomized, placebo-controlled, double-blind, phase I/II trial of 98 patients with platelet counts less than 30 × 109 /L as a result of AML or MDS. No significant improvements in platelet counts were recorded. Significant hemorrhage was reported in ten (16%) patients given eltrombopag and nine (26%) patients given placebo. No difference in disease progression or overall survival was observed.[14]
Eltrombopag appeared to hasten platelet recovery and reduce the number of platelet transfusions needed when added in an unblinded fashion to induction chemotherapy in older FLT3-negative AML patients.[15] However, in a separate, randomized double-blind study of 148 patients, eltrombopag or placebo was added to high-dose induction chemotherapy.[16] The results of this study did not indicate any clinical benefit of eltrombopag over placebo. Given the minimal efficacy signal at this point, eltrombopag is not routinely recommended in the supportive care or remission induction setting.
Antimicrobial therapy
Empiric broad spectrum antimicrobial therapy is an absolute necessity for febrile patients who are profoundly neutropenic.[17,18] Careful instruction in personal hand hygiene, dental care, and recognition of early signs of infection are appropriate in all patients. Elaborate isolation facilities (including filtered air, sterile food, and gut flora sterilization) are not indicated.[19,20] Likewise, there are no advantages to eating a cooked neutropenic diet, as demonstrated in randomized trials.[21]
Antibiotic prophylaxis with a fluoroquinolone and antifungal prophylaxis with an oral triazole or parenteral echinocandin is appropriate for patients with expected prolonged, profound neutropenia (<100/mm3 for 2 weeks for profound neutropenia lasting >7 days).[22] Unlike patients undergoing treatment for acute lymphoblastic lymphoma, Pneumocystis jirovecii prophylaxis is not routinely employed.
Nucleoside analog-based antiviral prophylaxis, such as acyclovir, is appropriate for patients who are seropositive for herpes simplex virus undergoing induction chemotherapy.[22]
References:
-
Rozovski U, Ohanian M, Ravandi F, et al.: Incidence of and risk factors for involvement of the central nervous system in acute myeloid leukemia. Leuk Lymphoma 56 (5): 1392-7, 2015.
-
Alakel N, Stölzel F, Mohr B, et al.: Symptomatic central nervous system involvement in adult patients with acute myeloid leukemia. Cancer Manag Res 9: 97-102, 2017.
-
Döhner H, Estey EH, Amadori S, et al.: Diagnosis and management of acute myeloid leukemia in adults: recommendations from an international expert panel, on behalf of the European LeukemiaNet. Blood 115 (3): 453-74, 2010.
-
Cassileth PA, Lynch E, Hines JD, et al.: Varying intensity of postremission therapy in acute myeloid leukemia. Blood 79 (8): 1924-30, 1992.
-
Supportive Care. In: Wiernik PH, Canellos GP, Dutcher JP, et al., eds.: Neoplastic Diseases of the Blood. 3rd ed. Churchill Livingstone, 1996, pp 779-967.
-
Slichter SJ: Controversies in platelet transfusion therapy. Annu Rev Med 31: 509-40, 1980.
-
Murphy MF, Metcalfe P, Thomas H, et al.: Use of leucocyte-poor blood components and HLA-matched-platelet donors to prevent HLA alloimmunization. Br J Haematol 62 (3): 529-34, 1986.
-
Rebulla P, Finazzi G, Marangoni F, et al.: The threshold for prophylactic platelet transfusions in adults with acute myeloid leukemia. Gruppo Italiano Malattie Ematologiche Maligne dell'Adulto. N Engl J Med 337 (26): 1870-5, 1997.
-
Leukocyte reduction and ultraviolet B irradiation of platelets to prevent alloimmunization and refractoriness to platelet transfusions. The Trial to Reduce Alloimmunization to Platelets Study Group. N Engl J Med 337 (26): 1861-9, 1997.
-
Price TH, Boeckh M, Harrison RW, et al.: Efficacy of transfusion with granulocytes from G-CSF/dexamethasone-treated donors in neutropenic patients with infection. Blood 126 (18): 2153-61, 2015.
-
Geller RB: Use of cytokines in the treatment of acute myelocytic leukemia: a critical review. J Clin Oncol 14 (4): 1371-82, 1996.
-
Rowe JM, Andersen JW, Mazza JJ, et al.: A randomized placebo-controlled phase III study of granulocyte-macrophage colony-stimulating factor in adult patients (> 55 to 70 years of age) with acute myelogenous leukemia: a study of the Eastern Cooperative Oncology Group (E1490). Blood 86 (2): 457-62, 1995.
-
Stone RM, Berg DT, George SL, et al.: Granulocyte-macrophage colony-stimulating factor after initial chemotherapy for elderly patients with primary acute myelogenous leukemia. Cancer and Leukemia Group B. N Engl J Med 332 (25): 1671-7, 1995.
-
Platzbecker U, Wong RS, Verma A, et al.: Safety and tolerability of eltrombopag versus placebo for treatment of thrombocytopenia in patients with advanced myelodysplastic syndromes or acute myeloid leukaemia: a multicentre, randomised, placebo-controlled, double-blind, phase 1/2 trial. Lancet Haematol 2 (10): e417-26, 2015.
-
Mukherjee S, Li H, Hobbs BP: A single arm, phase II study of eltrombopag to enhance platelet count recovery in older patients with acute myeloid leukemia (AML) undergoing remission induction therapy. [Abstract] Blood 134 (Suppl 1): 2595, 2019.
-
Frey N, Jang JH, Szer J, et al.: Eltrombopag treatment during induction chemotherapy for acute myeloid leukaemia: a randomised, double-blind, phase 2 study. Lancet Haematol 6 (3): e122-e131, 2019.
-
Hughes WT, Armstrong D, Bodey GP, et al.: From the Infectious Diseases Society of America. Guidelines for the use of antimicrobial agents in neutropenic patients with unexplained fever. J Infect Dis 161 (3): 381-96, 1990.
-
Rubin M, Hathorn JW, Pizzo PA: Controversies in the management of febrile neutropenic cancer patients. Cancer Invest 6 (2): 167-84, 1988.
-
Armstrong D: Symposium on infectious complications of neoplastic disease (Part II). Protected environments are discomforting and expensive and do not offer meaningful protection. Am J Med 76 (4): 685-9, 1984.
-
Sherertz RJ, Belani A, Kramer BS, et al.: Impact of air filtration on nosocomial Aspergillus infections. Unique risk of bone marrow transplant recipients. Am J Med 83 (4): 709-18, 1987.
-
Gardner A, Mattiuzzi G, Faderl S, et al.: Randomized comparison of cooked and noncooked diets in patients undergoing remission induction therapy for acute myeloid leukemia. J Clin Oncol 26 (35): 5684-8, 2008.
-
Taplitz RA, Kennedy EB, Bow EJ, et al.: Antimicrobial Prophylaxis for Adult Patients With Cancer-Related Immunosuppression: ASCO and IDSA Clinical Practice Guideline Update. J Clin Oncol 36 (30): 3043-3054, 2018.
Treatment of Newly Diagnosed AML
Treatment Options for Newly Diagnosed (Untreated; Remission Induction) AML
Treatment options for newly diagnosed (untreated; remission induction) acute myeloid leukemia (AML) include the following:
- Chemotherapy.
Chemotherapy
Chemotherapy for AML is divided into the following two general categories:
- Intensive remission-induction chemotherapy.
- Nonintensive chemotherapy.
One of the following combination chemotherapy regimens may be used as intensive remission induction therapy:
- Cytarabine plus daunorubicin.[1,2]
- Cytarabine plus idarubicin.[3,4,5,6]
- Cytarabine plus mitoxantrone.[7]
- Cytarabine plus anthracycline plus midostaurin.[8]
- Cytarabine plus anthracycline plus gemtuzumab ozogamicin.[9]
- Liposomal daunorubicin-cytarabine (CPX-351).[10]
- Intrathecal cytarabine or methotrexate may be used to treat central nervous system (CNS) leukemia, if present.
The two-drug regimen of cytarabine given as a continuous infusion for 7 days and a 3-day course of anthracycline (the so-called 7 + 3 induction therapy) results in a complete response rate of approximately 65%. In most instances, there is no further clinical benefit when adding potentially non-cross−resistant drugs (such as fludarabine, topoisomerase inhibitors, thioguanine, mitoxantrone, histone deacetylases inhibitors, or clofarabine) to a 7 + 3 regimen. Cladribine, when added to 7 + 3 induction chemotherapy, showed improved remission rates [11] and survival rates [12] across two randomized controlled trials, but this regimen has not been widely adopted in the absence of confirmatory trials. The addition of midostaurin and gemtuzumab ozogamicin to intensive induction chemotherapy is discussed below.
The choice of anthracycline and the dose-intensity of anthracycline may influence the survival of patients with AML. Idarubicin appeared to be more effective than daunorubicin, particularly in younger adults, although the doses of idarubicin and daunorubicin may not have been equivalent.[3,4,5,6] No significant survival difference between daunorubicin and mitoxantrone has been reported.[13]
Selection of an anthracycline
At present, there is no conclusive evidence to recommend one anthracycline over another.
Evidence (anthracyclines):
- In a systematic review and meta-analysis, 18 randomized controlled trials that included 6,755 patients assessed the use of idarubicin versus daunorubicin as part of induction chemotherapy.[14]
- The use of idarubicin led to improved outcomes, including overall survival (OS), when compared with daunorubicin (12 studies, 5,976 patients; hazard ratio [HR], 0.90; 95% confidence interval [CI], 0.84−0.96; P = .0008). However, there was an increased risk of death during induction (14 studies, 6,349 patients; relative risk [RR], 1.18; 95% CI, 1.01−1.36; P = .03) and mucositis (five studies, 2,000 patients; RR, 1.22; 95% CI, 1.04−1.44; P = .02) with idarubicin as compared with daunorubicin. Moreover, the survival benefit for idarubicin was no longer present if a daunorubicin dose of at least 180 mg/m2 was used (four studies, 2,867 patients; HR, 0.91; 95% CI, 0.82−1.00; P = .06).
- In patients aged 60 years and younger, outcomes for those who received daunorubicin (90 mg/m2 /dose, total induction dosing at 270 mg/m2) were superior to those who received more traditional dosing (45 mg/m2 /dose; total dose = 135 mg/m2). The complete remission (CR) rate was 71% versus 57% (P < .001), and the median survival was 24 months versus 16 months (P = .003).[15]
- No randomized comparison data between daunorubicin at 270 mg/m2 and daunorubicin at 180 mg/m2, or between daunorubicin at 270 mg/m2 and idarubicin, are available.
Addition of an FLT3 inhibitor
Mutations in the tyrosine kinase domain (TKD) and internal tandem duplications (ITD) of the FLT3 gene are frequent in AML and are often associated with an inferior outcome.
Midostaurin
Evidence (midostaurin):
- A multicenter, randomized, phase III trial (NCT00651261) included patients with FLT3-mutated AML. Patients received either the FLT3/multikinase inhibitor, midostaurin, or placebo in addition to cytarabine and daunorubicin induction chemotherapy. The addition of midostaurin led to improved survival (median, 75 vs. 26 months; HR for death, 0.78; one-sided P = .009).[8]
- The event-free survival (defined as the time from randomization to relapse, death from any cause, or failure to achieve protocol-specified CR) was improved for patients in the midostaurin arm (HR for event or death, 0.78; one-sided P = .002), and the survival benefit was consistent across all FLT3 mutation subtypes. The rates of CR (59% vs. 54%) and time to neutrophil count recovery were similar between the two arms.[8][Level of evidence A1]
The U.S. Food and Drug Administration (FDA) approved midostaurin in combination with induction therapy for patients with AML and any FLT3 mutation.
Quizartinib
Evidence (quizartinib):
- A multicenter, randomized, phase III trial (NCT02668653) included patients with FLT3-ITD mutated AML. Patients received either the selective ITD-specific FLT3 inhibitor, quizartinib, or placebo in addition to cytarabine and daunorubicin induction chemotherapy. The addition of quizartinib led to improved survival (median, 31.9 vs. 15.1 months; HR for death, 0.78; P = .032).[16]
- The EFS (defined as the time from randomization to lack of CR within 42 days from the start of the last induction cycle, relapse, or death from any cause, whichever occurred first) was similar for patients in the quizartinib and placebo arms (HR for event or death, 0.92; 95% CI, 0.75–1.11; P = .24). The rates of CR and time to neutrophil count recovery were similar between the two arms.[16][Level of evidence A1]
The FDA approved quizartinib in combination with induction therapy for patients with AML and an FLT3-ITD mutation but not for patients with other FLT3 mutations, such as FLT3-TKD.
The addition of an FLT3 inhibitor to induction chemotherapy is the standard of care for patients with FLT3-mutated AML who are eligible for intensive chemotherapy. An ongoing study (NCT03836209) is evaluating which FLT3 inhibitor is best for patients with FLT3-ITD AML receiving up-front chemotherapy. Additional studies are evaluating FLT3 inhibitors in combination with hypomethylating agents and venetoclax in patients who are not candidates for intensive therapy.
Addition of gemtuzumab ozogamicin
Evidence (gemtuzumab ozogamicin):
- In a meta-analysis of more than 3,000 patients, the addition of the CD33-directed immunotoxin gemtuzumab ozogamicin to cytarabine plus anthracycline or clofarabine plus anthracycline led to a small increase in the OS rate at 5 years (30.7% vs. 34.6%; HR, 0.90; 95% CI, 0.82−0.98; P = .01).[9]
- The improvement in the 5-year OS rate was seen across all ages, but this effect was greatest in patients with favorable-risk cytogenetics (55.2% vs. 76.3%; HR, 0.47; 95% CI, 0.31−0.73; P = .0005), and to a lesser extent with intermediate-risk cytogenetics (34.1% vs. 39.4%; HR, 0.84; 95% CI, 0.75−0.95; P = .007). It was not seen in patients with an adverse-risk karyotype.[9][Level of evidence A1]
- In contrast, gemtuzumab ozogamicin did not improve the 1-year survival rate of older patients who received low-dose cytarabine, although the CR rate increased from 17% to 30% (odds ratio [OR], 0.48; 95% CI, 0.32–0.73; P = .006).[17]
The FDA label for gemtuzumab ozogamicin includes a boxed warning about the risk of hepatotoxicity, including severe or fatal hepatic sinusoidal obstruction syndrome.
Liposomal daunorubicin-cytarabine (CPX-351)
CPX-351 is a two-drug liposomal encapsulation that delivers cytarabine and daunorubicin at a fixed 5:1 synergistic molar ratio.
Evidence (CPX-351):
- A multicenter trial investigated CPX-351 in patients aged 60 to 75 years with therapy-related AML, AML with a history of myelodysplastic syndrome (MDS), or AML with myelodysplasia-related changes.[10]
- Compared with 7 + 3 induction chemotherapy, CPX-351 resulted in a better overall remission rate (47.7% vs. 33.3%; P = .016), and improved median OS (9.56 vs. 5.95 months; HR, 0.69; 95% CI, 0.52−0.90; P = .003).[10][Level of evidence A1]
- The rates of early mortality and toxicities were similar between the two arms. However, the median time to recovery of neutrophils and platelets was longer for CPX-351 (35.0 and 36.5 days, respectively) as compared with 7 + 3 induction chemotherapy (29 and 29 days, respectively).
Older adults or adults with significant comorbid conditions
Some patients may decline or be too frail for intensive induction chemotherapy. Low-dose cytarabine, decitabine, azacitidine, or best supportive care can be considered equivalently effective treatment approaches for older patients with AML who decline traditional 7 + 3 induction chemotherapy. Unlike a succinct course of 7 + 3 induction, these less-intensive therapies are continued indefinitely, as long as the patient is deriving benefit (i.e., until disease progression or significant toxicity occurs).
One of the following chemotherapy regimens may be used as less-intensive therapy:
- Hypomethylating agents (azacitidine and decitabine).
- Low-dose cytarabine.
- Venetoclax plus hypomethylating agents or low-dose cytarabine.
- Glasdegib plus low-dose cytarabine.
- Ivosidenib with or without azacitidine.
- Enasidenib.
- Intrathecal cytarabine or methotrexate may be used to treat CNS leukemia, if present.
Evidence (chemotherapy for patients who decline intensive remission induction therapy):
- Hypomethylating agents: The hypomethylating agents azacitidine and decitabine are used commonly in this population of older patients, particularly in the United States. Although approval of the drugs by the FDA is for an MDS indication, the registration studies leading to approval included patients with 20% to 30% myeloblasts, or what would now be considered oligoblastic AML.[18,19]
- Azacitidine: An international phase III trial (NCT01074047) randomly assigned patients with AML who were 65 years or older to receive either azacitidine or conventional regimens (7 + 3 induction, low-dose cytarabine, or best supportive care alone).[20]
- Azacitidine led to a median OS of 10.4 months (95% CI, 8.0−12.7) as compared with 6.5 months (95% CI, 5.0−8.6) with conventional regimens (HR, 0.85; 95% CI, 0.69−1.03; P = .1009).[20][Level of evidence A1]
- Decitabine: One phase III trial randomly assigned 485 patients with AML who were older than 65 years to receive either decitabine (n = 242) or their preferred choice (n = 243) of either supportive care (n = 28) or low-dose cytarabine (n = 215).[21]
- Although rates of CR + CRp (CR with incomplete platelet recovery) were more than double in the decitabine arm (17.8%) compared with the treatment-choice arm (7.8%) (P = .001), median OS was not significantly improved for patients receiving decitabine (7.7 months) compared with the treatment of choice (5.0 months) (HR for death for decitabine, 0.85; 95% CI, .69–1.04; P = .11).
Compared with treatment for 5 consecutive days, treatment for 10 consecutive days may lead to higher response rates, particularly in those with TP53 mutations and/or unfavorable cytogenetic features.[22][Level of evidence C3]
- Low-dose cytarabine: Older adults who decline intensive remission-induction therapy or are considered ineligible for intensive remission-induction therapy may derive benefit from low-dose cytarabine, administered twice daily for 10 days in cycles repeated every 4 to 6 weeks.[23]
- The CR rate using this regimen was 18% compared with 1% for patients treated with hydroxyurea (P = .006).
- Survival with low-dose cytarabine was better than was survival with hydroxyurea (OR, 0.60; 95% CI, 0.44–0.81; P = .009).[23][Level of evidence A1]
- Venetoclax plus hypomethylating agents or low-dose cytarabine: The FDA approved venetoclax, an inhibitor of the anti-apoptotic protein BCL2, in combination with low-dose cytarabine or a hypomethylating agent for the treatment of AML in patients aged 75 years or older, and those who cannot undergo 7 + 3 induction chemotherapy because of comorbidities. Approval was granted based on the results of two studies.
- The first study (NCT02203773) was a nonrandomized, open-label, phase Ib clinical trial of venetoclax in combination with azacitidine or decitabine.[24]
- In this study, 35 (61%; 95% CI, 47.6%−74.0%) of 57 patients had CR or CR with incomplete hematologic recovery (CRi). The median duration of response for all patients with a response (CR + CRi + partial remission [PR]) was 8.4 months (range, 4.7−11.7; n = 36).
- The second study (NCT02287233) was a phase I/IIb study of venetoclax in combination with low-dose cytarabine in 82 patients with newly diagnosed AML, including patients with previous exposure to a hypomethylating agent for an antecedent hematologic disorder such as MDS.[25]
- In the 82 patients enrolled in the study, the CR/CRi rate was 54% (95% CI, 42%−65%), with a median duration of remission of 8.1 months (95% CI, 5.3−14.9 months). The median OS for all patients, irrespective of response, was 10.1 months (95% CI, 5.7−14.2 months).
- The most common adverse events with venetoclax combinations are gastrointestinal symptoms and myelosuppression, which may require delays between cycles, growth factor support, or decreased duration of venetoclax administration per cycle. Additionally, appropriate prophylactic measures are typically taken to prevent tumor lysis syndrome.[25][Level of evidence C3]
- Glasdegib plus low-dose cytarabine: Glasdegib, an oral inhibitor of the hedgehog pathway, combined with low-dose cytarabine was compared with low-dose cytarabine alone in a randomized, phase II open-label study that included 116 patients with AML who were aged 75 years or older or who had severe comorbid conditions (cardiac disease, renal impairment, or Eastern Cooperative Oncology Group performance status 2).[26]
- Of the 78 patients with AML who received glasdegib plus low-dose cytarabine, 24% (n = 19) of patients had a CR or CRi compared with 5% (2 of 38) of patients who received low-dose cytarabine alone. The median OS was 8.3 months (80%; CI, 6.6–9.5) for patients who received glasdegib/low-dose cytarabine and 4.3 months (80%; CI, 2.9–4.9) for patients who received low-dose cytarabine alone in patients with AML (HR, 0.46; 80% CI, 0.35–0.62; P = .0002).[26][Level of evidence A1]
Similar to venetoclax, the FDA approved glasdegib in combination with low-dose cytarabine for the treatment of AML in patients aged 75 years or older or who are unable to receive intensive induction chemotherapy.
- Ivosidenib: For older or frail patients with AML that harbors an IDH1 mutation, the IDH1 inhibitor ivosidenib is an option. The FDA approved ivosidenib alone or in combination with azacitidine for the treatment of AML that has a susceptible IDH1 mutation (detected by an FDA-approved diagnostic test) in adults aged 75 years or older with newly diagnosed AML or who have comorbidities that preclude the use of intensive induction chemotherapy. Within the context of a phase I study, 34 patients with newly diagnosed IDH1-mutated AML who were not candidates for intensive induction chemotherapy were treated with single-agent ivosidenib.[27]
- Differentiation syndrome was reported in 6 patients (18%), but did not require treatment discontinuation.
- The rate of CR plus CRi was 42.4% (95% CI, 25.5%−60.8%), with a median duration of response that was not reached (lower bound of the 95% CI was 4.6 months).
- For all 34 patients on the study, the median OS was 12.6 months (95% CI, 4.5−25.7).[27][Level of evidence C3]
The combination of azacitidine and ivosidenib was evaluated in a double-blind, randomized, placebo-controlled, phase III trial in patients with newly diagnosed AML who were not eligible for intensive induction chemotherapy. The intention-to-treat analysis included 72 patients treated with azacitidine and ivosidenib and 74 patients treated with azacitidine and placebo. A supplemental new drug application for ivosidenib in combination with azacitidine for patients with untreated IDH1-mutated AML is under priority review with the FDA.[28]
- At a median follow-up of 12.4 months, the primary end point of EFS was improved in patients who received azacitidine and ivosidenib, compared with patients who received azacitidine and placebo (HR, 0.33; 95% CI, 0.16–0.69).
- The median OS was 24 months for patients who received azacitidine and ivosidenib and 7.9 months for patients who received azacitidine and placebo (HR for death, 0.44; 95% CI, 0.27–0.73).
- Differentiation syndrome occurred in 14% of patients who received azacitidine and ivosidenib and 8% of patients who received azacitidine and placebo. The incidence of bleeding events was 41% with azacitidine and ivosidenib and 29% with azacitidine and placebo. Infection of any grade was seen in 28% of patients who received azacitidine and ivosidenib and 49% of patients who received azacitidine and placebo. For more information about differentiation syndrome, see the Treatment of Newly Diagnosed Acute Promyelocytic Leukemia section.[28][Level of evidence A1]
- Enasidenib: For patients with an AML that harbors a mutation in IDH2, the IDH2 inhibitor enasidenib is an option for older or frail patients. Although it does not have approval in this setting, enasidenib monotherapy was evaluated in a phase I/II trial in patients with newly diagnosed AML who were not eligible for standard chemotherapy.[29]
- Of the 39 patients enrolled, seven (18%) had CR, one (3%) had CRi, and two (5%) had a PR.
- Median OS for all patients was 11.3 months (95% CI, 5.7−15.1), and was not reached for patients who had a response.[29][Level of evidence C3]
Current Clinical Trials
Use our advanced clinical trial search to find NCI-supported cancer clinical trials that are now enrolling patients. The search can be narrowed by location of the trial, type of treatment, name of the drug, and other criteria. General information about clinical trials is also available.
References:
-
Yates J, Glidewell O, Wiernik P, et al.: Cytosine arabinoside with daunorubicin or adriamycin for therapy of acute myelocytic leukemia: a CALGB study. Blood 60 (2): 454-62, 1982.
-
Dillman RO, Davis RB, Green MR, et al.: A comparative study of two different doses of cytarabine for acute myeloid leukemia: a phase III trial of Cancer and Leukemia Group B. Blood 78 (10): 2520-6, 1991.
-
Wiernik PH, Banks PL, Case DC, et al.: Cytarabine plus idarubicin or daunorubicin as induction and consolidation therapy for previously untreated adult patients with acute myeloid leukemia. Blood 79 (2): 313-9, 1992.
-
Vogler WR, Velez-Garcia E, Weiner RS, et al.: A phase III trial comparing idarubicin and daunorubicin in combination with cytarabine in acute myelogenous leukemia: a Southeastern Cancer Study Group Study. J Clin Oncol 10 (7): 1103-11, 1992.
-
Berman E, Heller G, Santorsa J, et al.: Results of a randomized trial comparing idarubicin and cytosine arabinoside with daunorubicin and cytosine arabinoside in adult patients with newly diagnosed acute myelogenous leukemia. Blood 77 (8): 1666-74, 1991.
-
Mandelli F, Petti MC, Ardia A, et al.: A randomised clinical trial comparing idarubicin and cytarabine to daunorubicin and cytarabine in the treatment of acute non-lymphoid leukaemia. A multicentric study from the Italian Co-operative Group GIMEMA. Eur J Cancer 27 (6): 750-5, 1991.
-
Löwenberg B, Suciu S, Archimbaud E, et al.: Mitoxantrone versus daunorubicin in induction-consolidation chemotherapy--the value of low-dose cytarabine for maintenance of remission, and an assessment of prognostic factors in acute myeloid leukemia in the elderly: final report. European Organization for the Research and Treatment of Cancer and the Dutch-Belgian Hemato-Oncology Cooperative Hovon Group. J Clin Oncol 16 (3): 872-81, 1998.
-
Stone RM, Mandrekar SJ, Sanford BL, et al.: Midostaurin plus Chemotherapy for Acute Myeloid Leukemia with a FLT3 Mutation. N Engl J Med 377 (5): 454-464, 2017.
-
Hills RK, Castaigne S, Appelbaum FR, et al.: Addition of gemtuzumab ozogamicin to induction chemotherapy in adult patients with acute myeloid leukaemia: a meta-analysis of individual patient data from randomised controlled trials. Lancet Oncol 15 (9): 986-96, 2014.
-
Lancet JE, Uy GL, Cortes JE, et al.: CPX-351 (cytarabine and daunorubicin) Liposome for Injection Versus Conventional Cytarabine Plus Daunorubicin in Older Patients With Newly Diagnosed Secondary Acute Myeloid Leukemia. J Clin Oncol 36 (26): 2684-2692, 2018.
-
Holowiecki J, Grosicki S, Robak T, et al.: Addition of cladribine to daunorubicin and cytarabine increases complete remission rate after a single course of induction treatment in acute myeloid leukemia. Multicenter, phase III study. Leukemia 18 (5): 989-97, 2004.
-
Holowiecki J, Grosicki S, Giebel S, et al.: Cladribine, but not fludarabine, added to daunorubicin and cytarabine during induction prolongs survival of patients with acute myeloid leukemia: a multicenter, randomized phase III study. J Clin Oncol 30 (20): 2441-8, 2012.
-
Arlin Z, Case DC, Moore J, et al.: Randomized multicenter trial of cytosine arabinoside with mitoxantrone or daunorubicin in previously untreated adult patients with acute nonlymphocytic leukemia (ANLL). Lederle Cooperative Group. Leukemia 4 (3): 177-83, 1990.
-
Li X, Xu S, Tan Y, et al.: The effects of idarubicin versus other anthracyclines for induction therapy of patients with newly diagnosed leukaemia. Cochrane Database Syst Rev (6): CD010432, 2015.
-
Fernandez HF, Sun Z, Yao X, et al.: Anthracycline dose intensification in acute myeloid leukemia. N Engl J Med 361 (13): 1249-59, 2009.
-
Erba HP, Montesinos P, Kim HJ, et al.: Quizartinib plus chemotherapy in newly diagnosed patients with FLT3-internal-tandem-duplication-positive acute myeloid leukaemia (QuANTUM-First): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 401 (10388): 1571-1583, 2023.
-
Burnett AK, Hills RK, Hunter AE, et al.: The addition of gemtuzumab ozogamicin to low-dose Ara-C improves remission rate but does not significantly prolong survival in older patients with acute myeloid leukaemia: results from the LRF AML14 and NCRI AML16 pick-a-winner comparison. Leukemia 27 (1): 75-81, 2013.
-
Silverman LR, Demakos EP, Peterson BL, et al.: Randomized controlled trial of azacitidine in patients with the myelodysplastic syndrome: a study of the cancer and leukemia group B. J Clin Oncol 20 (10): 2429-40, 2002.
-
Kantarjian H, O'brien S, Cortes J, et al.: Results of intensive chemotherapy in 998 patients age 65 years or older with acute myeloid leukemia or high-risk myelodysplastic syndrome: predictive prognostic models for outcome. Cancer 106 (5): 1090-8, 2006.
-
Dombret H, Seymour JF, Butrym A, et al.: International phase 3 study of azacitidine vs conventional care regimens in older patients with newly diagnosed AML with >30% blasts. Blood 126 (3): 291-9, 2015.
-
Kantarjian HM, Thomas XG, Dmoszynska A, et al.: Multicenter, randomized, open-label, phase III trial of decitabine versus patient choice, with physician advice, of either supportive care or low-dose cytarabine for the treatment of older patients with newly diagnosed acute myeloid leukemia. J Clin Oncol 30 (21): 2670-7, 2012.
-
Welch JS, Petti AA, Miller CA, et al.: TP53 and Decitabine in Acute Myeloid Leukemia and Myelodysplastic Syndromes. N Engl J Med 375 (21): 2023-2036, 2016.
-
Burnett AK, Milligan D, Prentice AG, et al.: A comparison of low-dose cytarabine and hydroxyurea with or without all-trans retinoic acid for acute myeloid leukemia and high-risk myelodysplastic syndrome in patients not considered fit for intensive treatment. Cancer 109 (6): 1114-24, 2007.
-
DiNardo CD, Pratz KW, Letai A, et al.: Safety and preliminary efficacy of venetoclax with decitabine or azacitidine in elderly patients with previously untreated acute myeloid leukaemia: a non-randomised, open-label, phase 1b study. Lancet Oncol 19 (2): 216-228, 2018.
-
Wei AH, Strickland SA, Hou JZ, et al.: Venetoclax Combined With Low-Dose Cytarabine for Previously Untreated Patients With Acute Myeloid Leukemia: Results From a Phase Ib/II Study. J Clin Oncol 37 (15): 1277-1284, 2019.
-
Cortes JE, Heidel FH, Hellmann A, et al.: Randomized comparison of low dose cytarabine with or without glasdegib in patients with newly diagnosed acute myeloid leukemia or high-risk myelodysplastic syndrome. Leukemia 33 (2): 379-389, 2019.
-
Roboz GJ, DiNardo CD, Stein EM, et al.: Ivosidenib induces deep durable remissions in patients with newly diagnosed IDH1-mutant acute myeloid leukemia. Blood 135 (7): 463-471, 2020.
-
Montesinos P, Recher C, Vives S, et al.: Ivosidenib and Azacitidine in IDH1-Mutated Acute Myeloid Leukemia. N Engl J Med 386 (16): 1519-1531, 2022.
-
Pollyea DA, Tallman MS, de Botton S, et al.: Enasidenib, an inhibitor of mutant IDH2 proteins, induces durable remissions in older patients with newly diagnosed acute myeloid leukemia. Leukemia 33 (11): 2575-2584, 2019.
Treatment of AML in Remission
Although individual patients with acute myeloid leukemia (AML) have been reported to have long disease-free survival (DFS) or cure with a single cycle of chemotherapy,[1] postremission therapy is always indicated in therapy that is planned with curative intent. In a small randomized study conducted by the Eastern Cooperative Oncology Group, all patients who did not receive postremission therapy experienced a relapse after a short median complete remission (CR) duration.[2]
Treatment options for AML in remission (postremission phase) include the following:
- Chemotherapy with short-term (3−4 cycles), relatively intensive chemotherapy with cytarabine-based regimens similar to standard induction clinical trials (postremission chemotherapy) and postremission chemotherapy with more dose-intensive cytarabine-based treatment.
- Maintenance therapy (longer-term therapy at lower doses) has not been shown to benefit AML patients in a number of studies, but two strategies have shown a benefit:
- Midostaurin in patients with FLT3-mutated AML.
- Oral azacitidine.
- Hematopoietic cell (bone marrow or stem cell) transplant.
- High-dose chemotherapy with autologous peripheral blood stem cell rescue.
- High-dose marrow-ablative or reduced-intensity therapy followed by allogeneic hematopoietic cell transplant (HCT).
Chemotherapy
Nontransplant postremission therapy using cytarabine-containing regimens has treatment-related death rates that are usually less than 10% to 20% and has reported long-term DFS rates from 20% to 50%.[3,4,5,6] The optimal doses, schedules, and duration of postremission chemotherapy have not been determined.
The standard postremission therapy for AML patients in remission is high-dose cytarabine; however, there exists some controversy about whether it benefits all younger AML patients in first complete response versus selected subgroups, such as those with core-binding factor abnormalities.[7,8,9,10,11] The duration of postremission therapy has ranged from one cycle [4,6] to four or more cycles.[3,5]
Evidence (chemotherapy):
- A large, randomized trial that compared three different cytarabine-containing postremission therapy regimens showed a clear benefit in survival to patients younger than 60 years who received high-dose cytarabine.[3]
- Intensification of cytarabine dose or duration of postremission chemotherapy with conventionally dosed cytarabine did not improve DFS or overall survival (OS) in patients aged 60 years or older, as evidenced in the Medical Research Council (MRC-LEUK-AML11) trial.[12,13]
Dose-intensive cytarabine-based chemotherapy can be complicated by severe neurological [14] and/or pulmonary toxic effects [15] and should be administered by physicians experienced in these regimens at centers that are equipped to treat potential complications. In a retrospective analysis of 256 patients who received high-dose bolus cytarabine at a single institution, the most powerful predictor of cytarabine neurotoxicity was renal insufficiency. The incidence of neurotoxicity was significantly greater in patients treated with twice daily doses of 3 g/m2 /dose when compared with 2 g/m2 /dose.
Maintenance Therapy
While a number of older studies have included longer-term therapy at lower doses (maintenance), there has been no convincing evidence that maintenance therapy provides prolonged DFS or OS. However, maintenance therapy with midostaurin or oral azacitidine may improve outcomes.
Midostaurin
Evidence (midostaurin):
Mutations in the tyrosine kinase domain and internal tandem duplications of the FLT3 gene are frequent in AML and are often associated with an inferior outcome.
- In the multicenter, randomized, phase III RATIFY trial (NCT00651261), the FLT3/multikinase inhibitor midostaurin plus cytarabine and daunorubicin induction chemotherapy was compared with placebo in patients with FLT3-mutated AML.[16][Level of evidence A1]
- The addition of the FLT3/multikinase inhibitor midostaurin to cytarabine and daunorubicin induction chemotherapy led to improved survival (median, 75 vs. 26 months; hazard ratio [HR] for death, 0.78; one-sided P = .009).
- Patients who remained in remission after completion of consolidation therapy entered a maintenance phase in which they received midostaurin or placebo, administered at a dose of 50 mg orally twice daily, for twelve 28-day cycles. A maintenance regimen was administered for the full 12 cycles in 120 patients (69 in the midostaurin group, and 51 in the placebo group).
- An ad-hoc landmark analysis from the end of maintenance revealed no difference in DFS between the two arms. Moreover, from the start of maintenance therapy there was no difference in OS between the two arms.[17]
While maintenance was well tolerated in the RATIFY study, only a small subset of patients tolerated midostaurin as maintenance therapy after chemotherapy or transplant in a separate phase II study.[18]
- In the multicenter, randomized phase II RADIUS trial (NCT01883362), the addition of midostaurin to standard of care was compared with standard of care alone after allogeneic HCT.[19] Similar to the RATIFY trial, midostaurin was administered at a dose of 50 mg orally twice daily, for twelve 28-day cycles. Thirty patients were enrolled in each arm, but only half were able to complete the entire 12 cycles of therapy.
- The estimated 18-month relapse-free survival (RFS) rate was 89% in the midostaurin arm and 76% in the standard of care arm (HR, 0.46; 95% confidence interval [CI], 0.12−1.86; P = 0.27). The predicted relative reduction in the risk of relapse with the addition of midostaurin was 54%.[19][Level of evidence B1]
Oral azacitidine
Evidence (oral azacitidine):
- The randomized, double-blind, placebo-controlled phase III QUAZAR AML-001 trial (NCT01757535) provided the basis for regulatory approval of oral azacitidine as maintenance therapy for AML.[20] The study included 472 patients with AML who were aged 55 years or older, were within 4 months of first CR or CR with incomplete hematologic recovery after induction chemotherapy with or without consolidation treatment, and were not candidates for allogeneic HCT. Oral azacitidine or placebo was administered at 300 mg daily for 14 days every 28 days. Treatment was continued until disease progression or unacceptable toxicity.
- For patients who received azacitidine, the median OS from randomization was 24.7 months (95% CI, 18.7−30.5) compared with 14.8 months (95% CI, 11.7−17.6) for patients receiving placebo (HR, 0.69; 95% CI, 0.55−0.86; P = .0009).[20][Level of evidence A1]
Hematopoietic Cell (Bone Marrow or Stem Cell) Transplant
Allogeneic HCT
Allogeneic HCT, even with minimal conditioning chemotherapy, results in the lowest incidence of leukemic relapse, even when compared with HCT from an identical twin (syngeneic HCT). This finding led to the concept of an immunologic graft-versus-leukemia effect, similar to (and related to) graft-versus-host disease. The improvement in freedom from relapse using allogeneic HCT as the primary postremission therapy is offset, at least in part, by the increased morbidity and mortality caused by graft-versus-host disease, veno-occlusive disease of the liver, and infection. The DFS rates using allogeneic transplant in first complete remission have ranged from 45% to 60%.[21,22,23,24]
The use of allogeneic HCT in adults requires either a human leukocyte antigen (HLA)-matched sibling donor, an HLA-matched unrelated donor, a haploidentical donor ("half HLA-matched"), or two well-matched umbilical cord blood units. Including patients who underwent HCT from 2007 to 2017, the 3-year probabilities of survival after HLA-matched sibling transplant were 59% (±1%) for patients with early disease, 53% (±1%) for patients with intermediate disease, and 29% (±1%) for patients with advanced disease, according to the Center for International Blood and Marrow Transplant Research registry.[24] The probabilities of survival after an unrelated donor transplant were 53% (±1%) for patients with early disease, 50% (±1%) for intermediate disease, and 27% (±1%) for patients with advanced disease. [Level of evidence C1]
Because HCT can cure more than 30% of patients who experience relapse after chemotherapy, some investigators suggested that allogeneic bone marrow transplant (BMT) can be reserved for early first relapse or second CR without compromising the number of patients who are ultimately cured.[25] Clinical and cytogenetic information can define certain subsets of patients with predictable better or worse prognoses according to favorable- and adverse-risk factors in those using postremission chemotherapy.[26]
- Favorable-risk AML: Favorable-risk factors include t(8;21), inv(16), normal karyotype with NPM1 mutation (in absence of FLT3 mutation), and normal karyotype with double cytosine-cytosine-adenosine-adenosine-thymidine (CCAAT)-enhancer binding protein (C/EBP)-alpha mutations. Patients in the favorable-risk group have a reasonable chance of cure with intensive postremission chemotherapy, and it may be reasonable to defer transplant in that group until early first relapse.
- Adverse-risk AML: Adverse-risk factors include deletion of 5q, 7q, 17p, inversion of 3 or t(3;3), abnormality of (17p), trisomy 8, t(6;9), t(9;22), most translocations involving chromosome 11q23, and mutations of the MLL gene, a complex or monosomal karyotype, a history of myelodysplasia or antecedent hematologic disorder, TP53 mutation, RUNX1 mutation, ASXL1 mutation, and normal karyotype with FLT3 mutation. The adverse-risk group is highly unlikely to be cured with postremission chemotherapy, and allogeneic BMT in first CR is a reasonable option for patients. However, even with allogeneic stem cell transplant, the outcome for patients with high-risk AML is poor (5-year DFS rate of 8% to 30% for patients with treatment-related leukemia or myelodysplasia).[27]
- Normal cytogenetics: Patients with normal cytogenetics are in an intermediate-risk group, and postremission management should be individualized based on additional molecular markers, patient comorbid factors, and patient preference or, preferably, managed according to a clinical trial.
A common clinical trial design used to evaluate the benefit of allogeneic transplant as consolidation therapy for AML in first remission is the so-called donor-no donor comparison. In this design, newly diagnosed AML patients who achieve a CR are deemed medically eligible for allogeneic transplant and undergo HLA typing. If a matched sibling or matched unrelated donor is identified, the patient is allocated to the transplant arm. Analysis of outcome is by intention to treat; that is, patients assigned to the donor arm who do not receive a transplant are grouped in the analysis with the patients who did actually receive a transplant. RFS is the usual end point for this type of trial. OS from the time of diagnosis is less frequently reported in these trials.
Investigators attempted to address this issue with a meta-analysis using data from 18 separate prospective trials of AML patients using the donor-no donor design, with data from an additional six trials included for sensitivity analysis.[28] The trials included in this meta-analysis enrolled adult patients aged 60 years and younger from 1982 to 2006. Median follow-up ranged from 42 months to 142 months. Preparative regimens were similar among the different trials. Allogeneic transplant was compared with autologous transplant (six trials) or with a variety of consolidation chemotherapy regimens, with high-dose cytarabine being the most common.
Most physicians who treat patients with leukemia agree that transplant should be offered to AML patients in first CR in the setting of adverse-risk cytogenetics and should not be offered to patients in first CR with favorable-risk cytogenetics.[26] However, older patients with favorable-risk AML who are unlikely to tolerate intensive cytarabine-based consolidation therapy can be considered for allogeneic HCT as postremission therapy.[29]
Autologous hematopoietic stem cell transplant
The role of autologous transplant for AML patients has diminished over time because of the improvements in the nonrelapse mortality associated with allogeneic HCT, as well as the advent of haploidentical and umbilical cord transplant expanding the potential donor pool so that nearly every patient has a donor.[30,31,32,33] Autologous HCT can yield DFS rates between 35% and 50% in patients with AML in first remission. Autologous HCT has also cured a smaller proportion of patients in second remission.[34,35,36,37,38,39,40] Treatment-related mortality rates of patients who have had autologous peripheral blood or marrow transplant range from 10% to 20%.
Current Clinical Trials
Use our advanced clinical trial search to find NCI-supported cancer clinical trials that are now enrolling patients. The search can be narrowed by location of the trial, type of treatment, name of the drug, and other criteria. General information about clinical trials is also available.
References:
-
Vaughan WP, Karp JE, Burke PJ: Long chemotherapy-free remissions after single-cycle timed-sequential chemotherapy for acute myelocytic leukemia. Cancer 45 (5): 859-65, 1980.
-
Cassileth PA, Harrington DP, Hines JD, et al.: Maintenance chemotherapy prolongs remission duration in adult acute nonlymphocytic leukemia. J Clin Oncol 6 (4): 583-7, 1988.
-
Mayer RJ, Davis RB, Schiffer CA, et al.: Intensive postremission chemotherapy in adults with acute myeloid leukemia. Cancer and Leukemia Group B. N Engl J Med 331 (14): 896-903, 1994.
-
Champlin R, Gajewski J, Nimer S, et al.: Postremission chemotherapy for adults with acute myelogenous leukemia: improved survival with high-dose cytarabine and daunorubicin consolidation treatment. J Clin Oncol 8 (7): 1199-206, 1990.
-
Rohatiner AZ, Gregory WM, Bassan R, et al.: Short-term therapy for acute myelogenous leukemia. J Clin Oncol 6 (2): 218-26, 1988.
-
Geller RB, Burke PJ, Karp JE, et al.: A two-step timed sequential treatment for acute myelocytic leukemia. Blood 74 (5): 1499-506, 1989.
-
Löwenberg B: Sense and nonsense of high-dose cytarabine for acute myeloid leukemia. Blood 121 (1): 26-8, 2013.
-
Weick JK, Kopecky KJ, Appelbaum FR, et al.: A randomized investigation of high-dose versus standard-dose cytosine arabinoside with daunorubicin in patients with previously untreated acute myeloid leukemia: a Southwest Oncology Group study. Blood 88 (8): 2841-51, 1996.
-
Löwenberg B, Pabst T, Vellenga E, et al.: Cytarabine dose for acute myeloid leukemia. N Engl J Med 364 (11): 1027-36, 2011.
-
Schaich M, Röllig C, Soucek S, et al.: Cytarabine dose of 36 g/m² compared with 12 g/m² within first consolidation in acute myeloid leukemia: results of patients enrolled onto the prospective randomized AML96 study. J Clin Oncol 29 (19): 2696-702, 2011.
-
Miyawaki S, Ohtake S, Fujisawa S, et al.: A randomized comparison of 4 courses of standard-dose multiagent chemotherapy versus 3 courses of high-dose cytarabine alone in postremission therapy for acute myeloid leukemia in adults: the JALSG AML201 Study. Blood 117 (8): 2366-72, 2011.
-
Stone RM, Berg DT, George SL, et al.: Postremission therapy in older patients with de novo acute myeloid leukemia: a randomized trial comparing mitoxantrone and intermediate-dose cytarabine with standard-dose cytarabine. Blood 98 (3): 548-53, 2001.
-
Goldstone AH, Burnett AK, Wheatley K, et al.: Attempts to improve treatment outcomes in acute myeloid leukemia (AML) in older patients: the results of the United Kingdom Medical Research Council AML11 trial. Blood 98 (5): 1302-11, 2001.
-
Baker WJ, Royer GL, Weiss RB: Cytarabine and neurologic toxicity. J Clin Oncol 9 (4): 679-93, 1991.
-
Haupt HM, Hutchins GM, Moore GW: Ara-C lung: noncardiogenic pulmonary edema complicating cytosine arabinoside therapy of leukemia. Am J Med 70 (2): 256-61, 1981.
-
Stone RM, Mandrekar SJ, Sanford BL, et al.: Midostaurin plus Chemotherapy for Acute Myeloid Leukemia with a FLT3 Mutation. N Engl J Med 377 (5): 454-464, 2017.
-
Larson RA, Mandrekar SJ, Sanford BL: An Analysis of Maintenance Therapy and Post-Midostaurin Outcomes in the International Prospective Randomized, Placebo-Controlled, Double-Blind Trial (CALGB 10603/RATIFY [Alliance]) for Newly Diagnosed Acute Myeloid Leukemia (AML) Patients with FLT3 Mutations. [Abstract] Blood 130 (Suppl 1):145, 2017.
-
Schlenk RF, Weber D, Fiedler W, et al.: Midostaurin added to chemotherapy and continued single-agent maintenance therapy in acute myeloid leukemia with FLT3-ITD. Blood 133 (8): 840-851, 2019.
-
Maziarz RT, Levis M, Patnaik MM, et al.: Midostaurin after allogeneic stem cell transplant in patients with FLT3-internal tandem duplication-positive acute myeloid leukemia. Bone Marrow Transplant 56 (5): 1180-1189, 2021.
-
Wei AH, Döhner H, Pocock C, et al.: Oral Azacitidine Maintenance Therapy for Acute Myeloid Leukemia in First Remission. N Engl J Med 383 (26): 2526-2537, 2020.
-
Clift RA, Buckner CD, Thomas ED, et al.: The treatment of acute non-lymphoblastic leukemia by allogeneic marrow transplantation. Bone Marrow Transplant 2 (3): 243-58, 1987.
-
Reiffers J, Gaspard MH, Maraninchi D, et al.: Comparison of allogeneic or autologous bone marrow transplantation and chemotherapy in patients with acute myeloid leukaemia in first remission: a prospective controlled trial. Br J Haematol 72 (1): 57-63, 1989.
-
Bostrom B, Brunning RD, McGlave P, et al.: Bone marrow transplantation for acute nonlymphocytic leukemia in first remission: analysis of prognostic factors. Blood 65 (5): 1191-6, 1985.
-
D'Souza A, Fretham C, Lee SJ, et al.: Current Use of and Trends in Hematopoietic Cell Transplantation in the United States. Biol Blood Marrow Transplant 26 (8): e177-e182, 2020.
-
Schiller GJ, Nimer SD, Territo MC, et al.: Bone marrow transplantation versus high-dose cytarabine-based consolidation chemotherapy for acute myelogenous leukemia in first remission. J Clin Oncol 10 (1): 41-6, 1992.
-
Döhner H, Estey E, Grimwade D, et al.: Diagnosis and management of AML in adults: 2017 ELN recommendations from an international expert panel. Blood 129 (4): 424-447, 2017.
-
Witherspoon RP, Deeg HJ, Storer B, et al.: Hematopoietic stem-cell transplantation for treatment-related leukemia or myelodysplasia. J Clin Oncol 19 (8): 2134-41, 2001.
-
Koreth J, Schlenk R, Kopecky KJ, et al.: Allogeneic stem cell transplantation for acute myeloid leukemia in first complete remission: systematic review and meta-analysis of prospective clinical trials. JAMA 301 (22): 2349-61, 2009.
-
Tallman MS, Wang ES, Altman JK, et al.: Acute Myeloid Leukemia, Version 3.2019, NCCN Clinical Practice Guidelines in Oncology. J Natl Compr Canc Netw 17 (6): 721-749, 2019.
-
Gerds AT, Appelbaum FR: To transplant or not to transplant for adult acute myeloid leukemia: an ever-evolving decision. Clin Adv Hematol Oncol 10 (10): 655-62, 2012.
-
Gooley TA, Chien JW, Pergam SA, et al.: Reduced mortality after allogeneic hematopoietic-cell transplantation. N Engl J Med 363 (22): 2091-101, 2010.
-
Ciurea SO, Zhang MJ, Bacigalupo AA, et al.: Haploidentical transplant with posttransplant cyclophosphamide vs matched unrelated donor transplant for acute myeloid leukemia. Blood 126 (8): 1033-40, 2015.
-
Ballen KK, Lazarus H: Cord blood transplant for acute myeloid leukaemia. Br J Haematol 173 (1): 25-36, 2016.
-
Chao NJ, Stein AS, Long GD, et al.: Busulfan/etoposide--initial experience with a new preparatory regimen for autologous bone marrow transplantation in patients with acute nonlymphoblastic leukemia. Blood 81 (2): 319-23, 1993.
-
Linker CA, Ries CA, Damon LE, et al.: Autologous bone marrow transplantation for acute myeloid leukemia using busulfan plus etoposide as a preparative regimen. Blood 81 (2): 311-8, 1993.
-
Sanz MA, de la Rubia J, Sanz GF, et al.: Busulfan plus cyclophosphamide followed by autologous blood stem-cell transplantation for patients with acute myeloblastic leukemia in first complete remission: a report from a single institution. J Clin Oncol 11 (9): 1661-7, 1993.
-
Cassileth PA, Andersen J, Lazarus HM, et al.: Autologous bone marrow transplant in acute myeloid leukemia in first remission. J Clin Oncol 11 (2): 314-9, 1993.
-
Jones RJ, Santos GW: Autologous bone marrow transplantation with 4-hydroperoxycyclophosphamide purging. In: Gale RP, ed.: Acute Myelogenous Leukemia: Progress and Controversies: Proceedings of a Wyeth-Ayerst-UCLA Symposia Western Workshop Held at Lake Lanier, Georgia, November 28-December 1, 1989. Wiley-Liss, 1990, pp 411-419.
-
Gorin NC, Aegerter P, Auvert B, et al.: Autologous bone marrow transplantation for acute myelocytic leukemia in first remission: a European survey of the role of marrow purging. Blood 75 (8): 1606-14, 1990.
-
Robertson MJ, Soiffer RJ, Freedman AS, et al.: Human bone marrow depleted of CD33-positive cells mediates delayed but durable reconstitution of hematopoiesis: clinical trial of MY9 monoclonal antibody-purged autografts for the treatment of acute myeloid leukemia. Blood 79 (9): 2229-36, 1992.
Treatment of Refractory or Recurrent AML
No standard treatment regimen exists for patients with refractory or recurrent acute myeloid leukemia (AML).[1,2]
Treatment options for refractory or recurrent AML include the following:
- Chemotherapy.
- Intensive salvage chemotherapy.
- Reduced-intensity therapy, including targeted therapy.
- Allogeneic hematopoietic cell transplant (HCT).
Chemotherapy
Intensive salvage chemotherapy
A number of intensive salvage chemotherapy regimens have demonstrated efficacy in recurrent AML, including the following:
Fludarabine, cytarabine, and filgrastim (FLAG)
FLAG has shown antileukemic activity in patients with relapsed and refractory AML.
Evidence (FLAG):
- A multicenter phase II study included 83 patients with relapsed or refractory AML or de novo refractory anemia with excess blasts in transformation. The primary end point was the rate of complete remission (CR) achieved after one or two courses of FLAG induction chemotherapy.[3]
- In patients with relapsed leukemia whose first remission lasted for 6 months or more, the CR rate was 81% (17 out of 21 patients).
- In patients with AML whose first remission lasted less than 6 months, or in patients with primary refractory disease, the CR rate was 30% (13 out of 44 patients).[3][Level of evidence C3]
- Myelosuppression and mucositis were common therapy-related toxicities.
Idarubicin has been added to this regimen as well (FLAG-Ida).[4]
Mitoxantrone, etoposide, and cytarabine (MEC)
Evidence (MEC):
- A study evaluated one course of MEC chemotherapy in 74 patients with AML and a poor prognosis. The population included 30 patients with relapsed AML, 28 patients with primary refractory AML, and 16 patients with secondary AML.[5]
- MEC demonstrated a CR induction rate of 55%.[5][Level of evidence C3]
- A phase III randomized Eastern Cooperative Oncology Group (ECOG) trial (E-2995) assessed 129 patients with one of the following disease statuses: relapsed AML less than 6 months after first CR; relapsed AML after allogeneic or autologous bone marrow transplant; second or greater AML relapse; primary induction failure; secondary AML; or high-risk myelodysplastic syndromes. Patients were randomly assigned to receive MEC with or without valspodar, a multidrug resistance modulator.
- The complete response rate was only 17% to 25% in both arms.[6][Level of evidence B3]
Standard or high-dose cytarabine and mitoxantrone
Evidence (standard or high-dose cytarabine and mitoxantrone):
- A study of cytarabine and mitoxantrone included 49 patients with relapsed or refractory AML.[7]
- Treatment was successful in 50% to 60% of patients who experienced relapse after initially obtaining a CR, with 62.5% of patients with AML in first relapse achieving M1 marrow.[7,8]
High-dose etoposide and cyclophosphamide
Evidence (high-dose etoposide and cyclophosphamide):
- Reported results have been similar to those for the combination of cytarabine and mitoxantrone.[9]
Idarubicin and cytarabine
Evidence (idarubicin and cytarabine):
- Reported results have been similar to those for the combination of cytarabine and mitoxantrone.[10,11]
Other intensive regimens
- High-dose cytarabine.[12]
- Cytarabine, daunorubicin, and etoposide (ADE).[13]
- Clofarabine plus cytarabine with or without filgrastim (CLAG).[14,15]
Reduced-intensity therapy, including targeted therapy
Patients who are unable or unwilling to undergo intensive therapy can be treated with reduced-intensity therapies, including the following:
Gilteritinib
Gilteritinib is an oral FLT3 inhibitor with activity in both internal tandem duplication (ITD) and tyrosine kinase domain (TKD) subtypes.
Evidence (gilteritinib):
- In a phase III trial, 371 patients were randomly assigned in a 2:1 ratio to receive either gilteritinib or preselected salvage chemotherapy (MEC, FLAG-Ida, azacitidine, or low-dose cytarabine).[16,17]
- With a median follow-up of 37.1 months, the median overall survival (OS) was 9.3 months for patients who received gilteritinib versus 5.6 months for patients who received chemotherapy (hazard ratio [HR] for death, 0.665; 95% confidence interval [CI], 0.52–0.85). The estimated OS rate at 2 years was 20.6% (95% CI, 15.8%–26.0%) for patients who received gilteritinib and 14.2% (95% CI, 8.3%–21.6%) for patients who received chemotherapy.[16] Among those treated with gilteritinib, the median duration of complete response or complete response with partial hematologic recovery was 10 months (interquartile range [IQR], 2.08–not evaluable), and 23 months (IQR, 4.9–not evaluable) for just those achieving a complete response.[17]
- The rates of complete response and complete response with partial hematologic recovery were higher in patients who received gilteritinib than in patients who received chemotherapy (34% vs. 15.3%; HR, 18.6; 95% CI, 9.8–27.4).
- Adverse events of grade 3 or higher were less common in patients who received gilteritinib when adjusted for exposure time.[16][Level of evidence A1] The most common adverse event of interest was increased liver transaminases for patients who received gilteritinib therapy. If liver transaminase levels (considered related to treatment) increase to more than 5 times the upper limit of normal, gilteritinib therapy is paused. When the enzymes return to less than 2.5 times the upper limit of normal, therapy can resume at a reduced dose of 80 mg once daily.[17][Level of evidence A1]
Enasidenib
Enasidenib is an oral small molecule inhibitor with activity against the mutant IDH2 enzyme.
Evidence (enasidenib):
- In a phase I/II study, 214 patients with relapsed or refractory AML were treated with enasidenib.[18]
- The overall response rate was 40.3% (95% CI, 33.0%–48.0%), with a median response duration of 5.8 months.
- This included 46 patients (26.1%) with a CR or a CR with incomplete hematologic recovery.
- Grade 3 or 4 treatment-related adverse events included indirect hyperbilirubinemia (12%) and differentiation syndrome (7%). As a result, the U.S. Food and Drug Administration (FDA) added a boxed warning to enasidenib, which alerts prescribers that differentiation syndrome can occur.[18][Level of evidence C3]
Ivosidenib
Ivosidenib is an oral small molecule inhibitor with activity against the mutant IDH1 enzyme.
Evidence (ivosidenib):
- In a phase I/II study, 179 patients with relapsed or refractory AML were treated with ivosidenib.[19]
- The overall response rate was 39.1% (95% CI, 31.9%–46.7%) with a median duration response of 6.5 months (range, 4.6–9.3). This included 54 patients (30.2%) with a CR or a CR with incomplete hematologic recovery.
- Over one-third of patients who initially required transfusions became transfusion independent.
- Grade 3 or higher prolongation of the QT interval was seen in 14 patients (7.8%), and grade 3 or higher differentiation was seen in 7 patients (3.9%). The FDA added a boxed warning to ivosidenib, which alerts prescribers that differentiation syndrome can occur.[19][Level of evidence C3]
Hypomethylating agents
Evidence (hypomethylating agents):
- In a retrospective analysis, 655 patients with relapsed or primary treatment-refractory AML received either azacitidine (57%) or decitabine (43%).[20]
- Overall, 16% of patients achieved a CR or a CR with incomplete hematologic recovery and experienced a median OS of 21 months.[20][Level of evidence C3]
Gemtuzumab ozogamicin
The antibody-targeted chemotherapy agent gemtuzumab ozogamicin has been evaluated in patients who had relapsed AML and expressed CD33.
Evidence (gemtuzumab ozogamicin):
- A pooled analysis of three open-label, single-arm, phase II studies included 277 patients.[21]
- The overall response rate was 26%. Thirty-five patients (13%) achieved a CR and 36 patients (13%) achieved a CR without platelet recovery. It is unclear whether the inadequate platelet recovery was the result of megakaryocyte toxic effects of gemtuzumab or subclinical residual leukemia. [21]
The long-term outcomes of patients who receive gemtuzumab and achieve CR without platelet recovery are unclear. Gemtuzumab induces profound bone marrow aplasia similar to leukemia induction chemotherapy and also has substantial hepatic toxic effects, including hepatic veno-occlusive disease.[21][Level of evidence C3]
Clofarabine with or without cytarabine
Evidence (clofarabine with or without cytarabine):
- Clofarabine, a purine nucleoside analogue, was studied as a single agent in 62 patients with relapsed or refractory AML.[22]
- Eight of 19 patients who received treatment for their first relapse experienced a complete response.
- Clofarabine was administered in combination with intermediate-dose cytarabine in patients with relapsed or refractory AML.[23]
- Seven of 29 patients with AML or myelodysplastic syndrome who received treatment for their first relapse experienced a CR.[23][Level of evidence C3]
Allogeneic Hematopoietic Cell Transplant
When patients with relapsed disease are treated aggressively, they may have extended disease-free survival (DFS); however, patients with relapsed disease can only be cured with HCT.[24][Level of evidence C2] Allogeneic HCT for patients in their second CR provides better DFS rates than transplant for patients in relapse.[25][Level of evidence C1]
Evidence (allogeneic HCT):
- Current transplant outcomes are reported by the Center for International Blood and Marrow Transplantation Registry. The group reported the following outcomes for transplants done in the United States between 2008 and 2018:[26][Level of evidence C2]
- The 3-year survival rate was 53% for patients with AML in second or subsequent CR versus 31% for patients with relapsed disease (or never in a CR) who underwent a matched sibling donor transplant.
- The 3-year survival rate was 50% for patients with AML in second or subsequent CR versus 30% for patients with relapsed disease (or who never achieved CR) who underwent a matched unrelated donor transplant.
Allogeneic HCT can be effective salvage therapy in some patients whose disease fails to go into remission with intensive chemotherapy (primary refractory leukemia). A number of retrospective studies have demonstrated the ability of allogeneic HCT to induce remission in primary refractory disease.[27]
Evidence (allogeneic HCT to induce remission):
- In one retrospective analysis of 168 patients with AML and primary refractory disease who underwent allogeneic HCT, the 5-year OS rate was 22%.[28]
- Another analysis was conducted among patients enrolled in a prospective cooperative group trial, SWOG S0106 (NCT00085709).[29]
- Of the 589 patients treated in the cooperative group trial, 150 (25%) had primary refractory disease.
- Among the 64 patients with primary refractory disease who received an allogeneic HCT, the 4-year survival rate was 48% compared with 4% among the 86 patients who did not undergo transplant.[29][Level of evidence C3]
Randomized trials testing the efficacy of this approach are not available.
Current Clinical Trials
Use our advanced clinical trial search to find NCI-supported cancer clinical trials that are now enrolling patients. The search can be narrowed by location of the trial, type of treatment, name of the drug, and other criteria. General information about clinical trials is also available.
References:
-
Döhner H, Estey E, Grimwade D, et al.: Diagnosis and management of AML in adults: 2017 ELN recommendations from an international expert panel. Blood 129 (4): 424-447, 2017.
-
Sekeres MA, Guyatt G, Abel G, et al.: American Society of Hematology 2020 guidelines for treating newly diagnosed acute myeloid leukemia in older adults. Blood Adv 4 (15): 3528-3549, 2020.
-
Jackson G, Taylor P, Smith GM, et al.: A multicentre, open, non-comparative phase II study of a combination of fludarabine phosphate, cytarabine and granulocyte colony-stimulating factor in relapsed and refractory acute myeloid leukaemia and de novo refractory anaemia with excess of blasts in transformation. Br J Haematol 112 (1): 127-37, 2001.
-
Pastore D, Specchia G, Carluccio P, et al.: FLAG-IDA in the treatment of refractory/relapsed acute myeloid leukemia: single-center experience. Ann Hematol 82 (4): 231-5, 2003.
-
Spadea A, Petti MC, Fazi P, et al.: Mitoxantrone, etoposide and intermediate-dose Ara-C (MEC): an effective regimen for poor risk acute myeloid leukemia. Leukemia 7 (4): 549-52, 1993.
-
Greenberg PL, Lee SJ, Advani R, et al.: Mitoxantrone, etoposide, and cytarabine with or without valspodar in patients with relapsed or refractory acute myeloid leukemia and high-risk myelodysplastic syndrome: a phase III trial (E2995). J Clin Oncol 22 (6): 1078-86, 2004.
-
Paciucci PA, Dutcher JP, Cuttner J, et al.: Mitoxantrone and ara-C in previously treated patients with acute myelogenous leukemia. Leukemia 1 (7): 565-7, 1987.
-
Hiddemann W, Kreutzmann H, Straif K, et al.: High-dose cytosine arabinoside and mitoxantrone: a highly effective regimen in refractory acute myeloid leukemia. Blood 69 (3): 744-9, 1987.
-
Brown RA, Herzig RH, Wolff SN, et al.: High-dose etoposide and cyclophosphamide without bone marrow transplantation for resistant hematologic malignancy. Blood 76 (3): 473-9, 1990.
-
Lambertenghi-Deliliers G, Maiolo AT, Annaloro C, et al.: Idarubicin in sequential combination with cytosine arabinoside in the treatment of relapsed and refractory patients with acute non-lymphoblastic leukemia. Eur J Cancer Clin Oncol 23 (7): 1041-5, 1987.
-
Harousseau JL, Reiffers J, Hurteloup P, et al.: Treatment of relapsed acute myeloid leukemia with idarubicin and intermediate-dose cytarabine. J Clin Oncol 7 (1): 45-9, 1989.
-
Herzig RH, Lazarus HM, Wolff SN, et al.: High-dose cytosine arabinoside therapy with and without anthracycline antibiotics for remission reinduction of acute nonlymphoblastic leukemia. J Clin Oncol 3 (7): 992-7, 1985.
-
Liu Yin JA, Wheatley K, Rees JK, et al.: Comparison of 'sequential' versus 'standard' chemotherapy as re-induction treatment, with or without cyclosporine, in refractory/relapsed acute myeloid leukaemia (AML): results of the UK Medical Research Council AML-R trial. Br J Haematol 113 (3): 713-26, 2001.
-
Becker PS, Kantarjian HM, Appelbaum FR, et al.: Clofarabine with high dose cytarabine and granulocyte colony-stimulating factor (G-CSF) priming for relapsed and refractory acute myeloid leukaemia. Br J Haematol 155 (2): 182-9, 2011.
-
Scappini B, Gianfaldoni G, Caracciolo F, et al.: Cytarabine and clofarabine after high-dose cytarabine in relapsed or refractory AML patients. Am J Hematol 87 (12): 1047-51, 2012.
-
Perl AE, Martinelli G, Cortes JE, et al.: Gilteritinib or Chemotherapy for Relapsed or Refractory FLT3-Mutated AML. N Engl J Med 381 (18): 1728-1740, 2019.
-
Perl AE, Larson RA, Podoltsev NA, et al.: Follow-up of patients with R/R FLT3-mutation-positive AML treated with gilteritinib in the phase 3 ADMIRAL trial. Blood 139 (23): 3366-3375, 2022.
-
Stein EM, DiNardo CD, Pollyea DA, et al.: Enasidenib in mutant IDH2 relapsed or refractory acute myeloid leukemia. Blood 130 (6): 722-731, 2017.
-
DiNardo CD, Stein EM, de Botton S, et al.: Durable Remissions with Ivosidenib in IDH1-Mutated Relapsed or Refractory AML. N Engl J Med 378 (25): 2386-2398, 2018.
-
Stahl M, DeVeaux M, Montesinos P, et al.: Hypomethylating agents in relapsed and refractory AML: outcomes and their predictors in a large international patient cohort. Blood Adv 2 (8): 923-932, 2018.
-
Larson RA, Sievers EL, Stadtmauer EA, et al.: Final report of the efficacy and safety of gemtuzumab ozogamicin (Mylotarg) in patients with CD33-positive acute myeloid leukemia in first recurrence. Cancer 104 (7): 1442-52, 2005.
-
Kantarjian H, Gandhi V, Cortes J, et al.: Phase 2 clinical and pharmacologic study of clofarabine in patients with refractory or relapsed acute leukemia. Blood 102 (7): 2379-86, 2003.
-
Faderl S, Gandhi V, O'Brien S, et al.: Results of a phase 1-2 study of clofarabine in combination with cytarabine (ara-C) in relapsed and refractory acute leukemias. Blood 105 (3): 940-7, 2005.
-
Forman SJ, Schmidt GM, Nademanee AP, et al.: Allogeneic bone marrow transplantation as therapy for primary induction failure for patients with acute leukemia. J Clin Oncol 9 (9): 1570-4, 1991.
-
Clift RA, Buckner CD, Thomas ED, et al.: The treatment of acute non-lymphoblastic leukemia by allogeneic marrow transplantation. Bone Marrow Transplant 2 (3): 243-58, 1987.
-
Phelan R, Arora M, Chen M: The US Summary Slides - HCT Trends and Survival Data. Center for International Blood and Marrow Transplant Research, 2020. Available online. Last accessed October 5, 2023.
-
Gyurkocza B, Lazarus HM, Giralt S: Allogeneic hematopoietic cell transplantation in patients with AML not achieving remission: potentially curative therapy. Bone Marrow Transplant 52 (8): 1083-1090, 2017.
-
Craddock C, Labopin M, Pillai S, et al.: Factors predicting outcome after unrelated donor stem cell transplantation in primary refractory acute myeloid leukaemia. Leukemia 25 (5): 808-13, 2011.
-
Othus M, Appelbaum FR, Petersdorf SH, et al.: Fate of patients with newly diagnosed acute myeloid leukemia who fail primary induction therapy. Biol Blood Marrow Transplant 21 (3): 559-64, 2015.
Treatment of Acute Promyelocytic Leukemia (APL)
Special consideration must be given to induction therapy for APL. Treatment is centered around the use of differentiating agents to clear the leukemic cells. Early mortality is related to bleeding, differentiation syndrome, or infection. High complete remission (CR) rates are very common across treatment regimens, and persistent disease or relapse is rare.
Treatment of Newly Diagnosed APL
Treatment options for newly diagnosed APL include the following:
- All-trans retinoic acid (ATRA) plus arsenic trioxide (ATO) for low- to intermediate-risk disease.
- ATRA plus chemotherapy, followed by ATO-based consolidation therapy for high-risk disease.
ATRA induces terminal differentiation of the leukemic cells followed by restoration of nonclonal hematopoiesis. Administration of ATRA leads to rapid resolution of coagulopathy in most patients, and heparin administration is not required in patients receiving ATRA. However, randomized trials have not shown a reduction in morbidity and mortality during ATRA induction when compared with chemotherapy. ATRA administration may result in the following conditions:
- Differentiation syndrome: Administration of ATRA can lead to a syndrome of respiratory distress, known as differentiation syndrome. Prompt recognition of the syndrome and aggressive administration of steroids can prevent severe respiratory distress.[1]
- Hyperleukocytosis: The optimal management of ATRA-induced hyperleukocytosis has not been established. Hyperleukocytosis in APL is typically treated with the addition of cytotoxic chemotherapy. Leukopheresis is not recommended because of an increased risk of complications such as bleeding events and disseminated intravascular coagulation.
Studies performed in the 1990s demonstrated that overall survival (OS) rates improved in patients receiving ATRA in addition to chemotherapy.[2,3] ATO, an agent with both differentiation-inducing and apoptosis-inducing properties against APL cells, is also used in the treatment of APL. Induction remission therapy for APL is determined by disease risk. Low- to intermediate-risk APL (white blood cell [WBC] count ≤10 × 109 /L) is treated without chemotherapy (ATRA and ATO), and high-risk is treated with a combination of ATRA and ATO plus chemotherapy.
ATRA plus ATO for low- to intermediate-risk disease
Evidence (ATRA plus ATO for low- to intermediate-risk disease):
- In a phase III, randomized controlled, multicenter trial in patients with APL classified as low-to-intermediate risk (WBC count, ≤10 × 109 /L) ATRA plus chemotherapy was compared with ATRA plus ATO.[4]
- CR rates were equally high for both groups. CR occurred in all 77 patients (100%) who could be evaluated in the ATRA plus ATO group and in 75 of 79 patients (95%) in the ATRA plus chemotherapy group (P = .12).
- With a median follow-up of 34.4 months, the 2-year OS rate was 99% (95% confidence interval [CI], 96%−100%) in the ATRA plus ATO group, and 91% (95% CI, 85%−97%) in the ATRA plus chemotherapy group (P = .02).
- The 2-year cumulative incidence of relapse was similarly low in both groups, 1% (95% CI, 0%−4%) in the ATRA plus ATO group and 6% (95% CI, 0%−11%) in the ATRA plus chemotherapy group (P = .24).
- The primary end point was event-free survival (EFS) (defined as no achievement of hematologic CR after induction therapy, no achievement of molecular CR after three consolidation courses, molecular relapse, hematologic relapse, or death). The 2-year EFS rate was 97% for the ATRA plus ATO group and 85% in the ATRA plus chemotherapy group (P < .001 for noninferiority).
- The results of this chemotherapy-free regimen for low- to intermediate-risk APL were confirmed in a second, phase III, randomized controlled trial.[5][Level of evidence A1]
ATRA plus chemotherapy, followed by ATO-based consolidation therapy for high-risk disease
Remission induction with a combination of anthracycline and ATRA is used for remission induction in patients with high-risk disease (WBC count, >10 × 109 /L).
Evidence (ATRA plus chemotherapy, followed by ATO-based consolidation therapy for high-risk disease):
- The AIDA-2000 study (NCT00180128) combined oral ATRA until CR or for a maximum of 45 days and four doses of intravenous idarubicin (12 mg/m2) on days 2, 4, 6, and 8. Consolidation was then risk stratified so low- to intermediate-risk patients received additional cycles of anthracycline and ATRA, and high-risk patients also received cytarabine, etoposide, 6-thioguanine, and CHT chemotherapy (6-mercaptopurine plus methotrexate) as consolidation.[6]
- After induction, 420 of 445 patients (94.4%) treated on the AIDA-2000 protocol were in CR. The 6-year OS rate was 87.4% and the cumulative incidence of relapse rate was 10.7%.[6][Level of evidence B4]
- In the phase II APML4 study, ATO was added to the ATRA-and-idarubicin remission−induction backbone.[7]
- Of the 124 patients who could be evaluated, there were 4 (3.2%) early deaths and 118 (95%) hematologic CRs.
- The 2-year freedom-from-relapse rate was 97.5% (95% CI, 90.4%−99.4%), the failure-free survival rate was 88.1% (95% CI, 80.7%−92.8%), and the OS rate was 93.2% (95% CI, 85.8%−96.8%).[7][Level of evidence B4]
An ATO-based regimen, which includes gemtuzumab ozogamicin as the only cytotoxic drug, has been developed.
- In a single-institution study, patients received ATRA plus ATO induction. They also received a dose of gemtuzumab ozogamicin (or idarubicin) if the WBC was greater than 10 × 109 /L on presentation or rose to more than 30 × 109 /L during induction.[8] Patients in remission received ATO and ATRA in alternating months for a total of seven cycles as consolidation; gemtuzumab ozogamicin was substituted if either ATO or ATRA were discontinued because of toxicity.
- Of the 54 patients with high-risk disease treated on the protocol, one patient received both gemtuzumab ozogamicin and idarubicin (12 mg/m2 daily for 3 days) because of persistent leukocytosis despite receiving gemtuzumab ozogamicin.
- The CR rate was 96%, and five (9%) patients relapsed.
- The 5-year rates were 81% for EFS, 89% for disease-free survival (DFS), and 86% for OS, indicating that the responses are durable.
- Across the entire study population, the 5-year OS rate was similar among the 45 high-risk patients who received gemtuzumab ozogamicin (84%) compared with the seven patients who received a dose of idarubicin (100%, P = .453).[8][Level of evidence B4]
Long-term follow up from this study has been published.[9]
It is important to note that most current regimens for the treatment of APL include some form of maintenance therapy. A meta-analysis of randomized trials has indicated that maintenance clearly improves DFS but not OS; however, these trials did not include ATO-containing regimens.
Treatment of Recurrent APL
Treatment options for recurrent APL include the following:
- ATO with or without chemotherapy.
- Hematopoietic cell transplant (HCT).
ATO with or without chemotherapy
ATO has high rates of second remission in patients with relapsed APL.[10] As a single agent, ATO can lead to complete response rates of 80% to 90% in patients with hematologic relapse, and 70% to 80% in patients with molecular remission.[11,12,13,14] The choice of salvage therapy is based on the previous therapy and interval of time between first remission and relapse.
- In patients with early relapse less than 6 months after ATRA and ATO, anthracycline-based regimens and ATRA as given for initial remission induction for high-risk disease should be considered.
- In patients who relapse less than 6 months after ATRA and anthracycline-based regimens (no previous ATO exposure), ATO-based regimens should be considered.
- In all patients with late relapse (>6 months), ATO-based regimens with or without anthracycline or gemtuzumab ozogamicin should be considered.
For patients receiving ATO as salvage therapy, a small randomized trial suggested that the addition of ATRA does not confer any benefit over ATO alone in patients who previously received ATRA.[14] In this 20-patient study, the complete response rate after one cycle of ATO with or without ATRA was 80%.
HCT
Some patients in second remission with ATO have experienced long-term DFS after autologous stem cell transplant,[15,16] and it can be considered in patients who are in molecular remission (negative quantitative polymerase chain reaction [PCR] on a marrow sample). Patients who do not go into remission or have evidence of measurable residual disease by quantitative PCR on a marrow sample after salvage therapy are considered for an allogeneic HCT.[17] A registry study reported a 3-year OS rate after transplant in second CR of 80% compared with 59% in patients without transplant (P = .03).[10]
Current Clinical Trials
Use our advanced clinical trial search to find NCI-supported cancer clinical trials that are now enrolling patients. The search can be narrowed by location of the trial, type of treatment, name of the drug, and other criteria. General information about clinical trials is also available.
References:
-
Frankel SR, Eardley A, Lauwers G, et al.: The "retinoic acid syndrome" in acute promyelocytic leukemia. Ann Intern Med 117 (4): 292-6, 1992.
-
Adès L, Guerci A, Raffoux E, et al.: Very long-term outcome of acute promyelocytic leukemia after treatment with all-trans retinoic acid and chemotherapy: the European APL Group experience. Blood 115 (9): 1690-6, 2010.
-
Sanz MA, Montesinos P, Vellenga E, et al.: Risk-adapted treatment of acute promyelocytic leukemia with all-trans retinoic acid and anthracycline monochemotherapy: long-term outcome of the LPA 99 multicenter study by the PETHEMA Group. Blood 112 (8): 3130-4, 2008.
-
Lo-Coco F, Avvisati G, Vignetti M, et al.: Retinoic acid and arsenic trioxide for acute promyelocytic leukemia. N Engl J Med 369 (2): 111-21, 2013.
-
Burnett AK, Russell NH, Hills RK, et al.: Arsenic trioxide and all-trans retinoic acid treatment for acute promyelocytic leukaemia in all risk groups (AML17): results of a randomised, controlled, phase 3 trial. Lancet Oncol 16 (13): 1295-305, 2015.
-
Lo-Coco F, Avvisati G, Vignetti M, et al.: Front-line treatment of acute promyelocytic leukemia with AIDA induction followed by risk-adapted consolidation for adults younger than 61 years: results of the AIDA-2000 trial of the GIMEMA Group. Blood 116 (17): 3171-9, 2010.
-
Iland HJ, Bradstock K, Supple SG, et al.: All-trans-retinoic acid, idarubicin, and IV arsenic trioxide as initial therapy in acute promyelocytic leukemia (APML4). Blood 120 (8): 1570-80; quiz 1752, 2012.
-
Ravandi F, Estey E, Jones D, et al.: Effective treatment of acute promyelocytic leukemia with all-trans-retinoic acid, arsenic trioxide, and gemtuzumab ozogamicin. J Clin Oncol 27 (4): 504-10, 2009.
-
Abaza Y, Kantarjian H, Garcia-Manero G, et al.: Long-term outcome of acute promyelocytic leukemia treated with all-trans-retinoic acid, arsenic trioxide, and gemtuzumab. Blood 129 (10): 1275-1283, 2017.
-
Lengfelder E, Lo-Coco F, Ades L, et al.: Arsenic trioxide-based therapy of relapsed acute promyelocytic leukemia: registry results from the European LeukemiaNet. Leukemia 29 (5): 1084-91, 2015.
-
Leoni F, Gianfaldoni G, Annunziata M, et al.: Arsenic trioxide therapy for relapsed acute promyelocytic leukemia: a bridge to transplantation. Haematologica 87 (5): 485-9, 2002.
-
Soignet SL, Frankel SR, Douer D, et al.: United States multicenter study of arsenic trioxide in relapsed acute promyelocytic leukemia. J Clin Oncol 19 (18): 3852-60, 2001.
-
Thirugnanam R, George B, Chendamarai E, et al.: Comparison of clinical outcomes of patients with relapsed acute promyelocytic leukemia induced with arsenic trioxide and consolidated with either an autologous stem cell transplant or an arsenic trioxide-based regimen. Biol Blood Marrow Transplant 15 (11): 1479-84, 2009.
-
Raffoux E, Rousselot P, Poupon J, et al.: Combined treatment with arsenic trioxide and all-trans-retinoic acid in patients with relapsed acute promyelocytic leukemia. J Clin Oncol 21 (12): 2326-34, 2003.
-
Yanada M, Tsuzuki M, Fujita H, et al.: Phase 2 study of arsenic trioxide followed by autologous hematopoietic cell transplantation for relapsed acute promyelocytic leukemia. Blood 121 (16): 3095-102, 2013.
-
Holter Chakrabarty JL, Rubinger M, Le-Rademacher J, et al.: Autologous is superior to allogeneic hematopoietic cell transplantation for acute promyelocytic leukemia in second complete remission. Biol Blood Marrow Transplant 20 (7): 1021-5, 2014.
-
de Botton S, Fawaz A, Chevret S, et al.: Autologous and allogeneic stem-cell transplantation as salvage treatment of acute promyelocytic leukemia initially treated with all-trans-retinoic acid: a retrospective analysis of the European acute promyelocytic leukemia group. J Clin Oncol 23 (1): 120-6, 2005.
Latest Updates to This Summary (03 / 06 / 2024)
The PDQ cancer information summaries are reviewed regularly and updated as new information becomes available. This section describes the latest changes made to this summary as of the date above.
Editorial changes were made to this summary.
This summary is written and maintained by the PDQ Adult Treatment Editorial Board, which is editorially independent of NCI. The summary reflects an independent review of the literature and does not represent a policy statement of NCI or NIH. More information about summary policies and the role of the PDQ Editorial Boards in maintaining the PDQ summaries can be found on the About This PDQ Summary and PDQ® Cancer Information for Health Professionals pages.
About This PDQ Summary
Purpose of This Summary
This PDQ cancer information summary for health professionals provides comprehensive, peer-reviewed, evidence-based information about the treatment of acute myeloid leukemia. It is intended as a resource to inform and assist clinicians in the care of their patients. It does not provide formal guidelines or recommendations for making health care decisions.
Reviewers and Updates
This summary is reviewed regularly and updated as necessary by the PDQ Adult Treatment Editorial Board, which is editorially independent of the National Cancer Institute (NCI). The summary reflects an independent review of the literature and does not represent a policy statement of NCI or the National Institutes of Health (NIH).
Board members review recently published articles each month to determine whether an article should:
- be discussed at a meeting,
- be cited with text, or
- replace or update an existing article that is already cited.
Changes to the summaries are made through a consensus process in which Board members evaluate the strength of the evidence in the published articles and determine how the article should be included in the summary.
The lead reviewer for Acute Myeloid Leukemia Treatment is:
- Aaron Gerds, MD (Cleveland Clinic Taussig Cancer Institute)
Any comments or questions about the summary content should be submitted to Cancer.gov through the NCI website's Email Us. Do not contact the individual Board Members with questions or comments about the summaries. Board members will not respond to individual inquiries.
Levels of Evidence
Some of the reference citations in this summary are accompanied by a level-of-evidence designation. These designations are intended to help readers assess the strength of the evidence supporting the use of specific interventions or approaches. The PDQ Adult Treatment Editorial Board uses a formal evidence ranking system in developing its level-of-evidence designations.
Permission to Use This Summary
PDQ is a registered trademark. Although the content of PDQ documents can be used freely as text, it cannot be identified as an NCI PDQ cancer information summary unless it is presented in its entirety and is regularly updated. However, an author would be permitted to write a sentence such as "NCI's PDQ cancer information summary about breast cancer prevention states the risks succinctly: [include excerpt from the summary]."
The preferred citation for this PDQ summary is:
PDQ® Adult Treatment Editorial Board. PDQ Acute Myeloid Leukemia Treatment. Bethesda, MD: National Cancer Institute. Updated <MM/DD/YYYY>. Available at: https://www.cancer.gov/types/leukemia/hp/adult-aml-treatment-pdq. Accessed <MM/DD/YYYY>. [PMID: 26389432]
Images in this summary are used with permission of the author(s), artist, and/or publisher for use within the PDQ summaries only. Permission to use images outside the context of PDQ information must be obtained from the owner(s) and cannot be granted by the National Cancer Institute. Information about using the illustrations in this summary, along with many other cancer-related images, is available in Visuals Online, a collection of over 2,000 scientific images.
Disclaimer
Based on the strength of the available evidence, treatment options may be described as either "standard" or "under clinical evaluation." These classifications should not be used as a basis for insurance reimbursement determinations. More information on insurance coverage is available on Cancer.gov on the Managing Cancer Care page.
Contact Us
More information about contacting us or receiving help with the Cancer.gov website can be found on our Contact Us for Help page. Questions can also be submitted to Cancer.gov through the website's Email Us.
Last Revised: 2024-03-06